

CMTM6 shapes antitumor T cell response through modulating protein expression of CD58 and PD-L1
- PMID: 37683639
- PMCID: PMC11113010
- DOI: 10.1016/j.ccell.2023.08.008
CMTM6 shapes antitumor T cell response through modulating protein expression of CD58 and PD-L1
Abstract
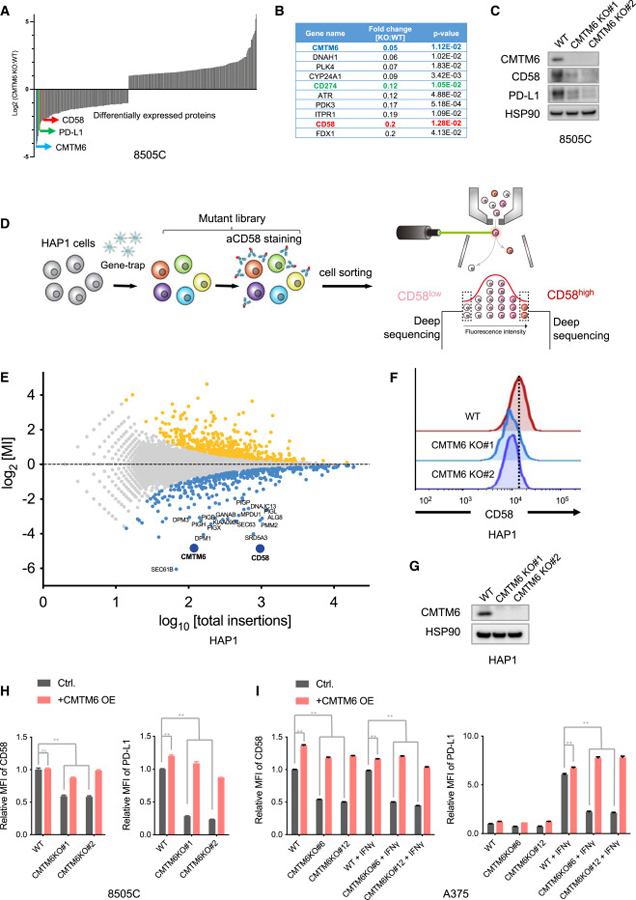
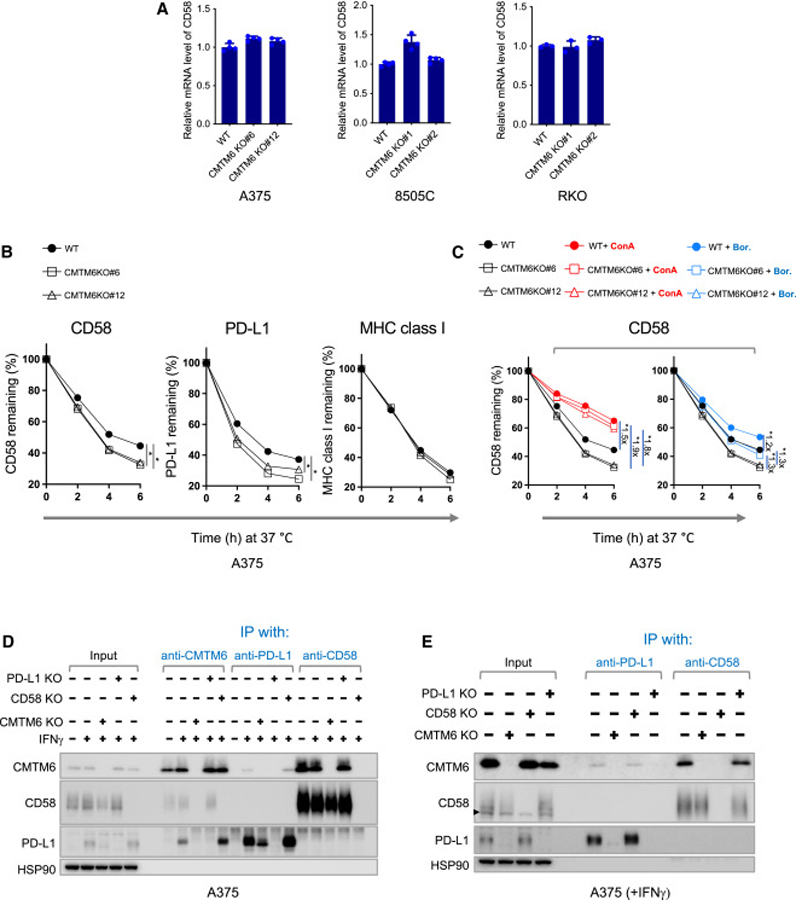
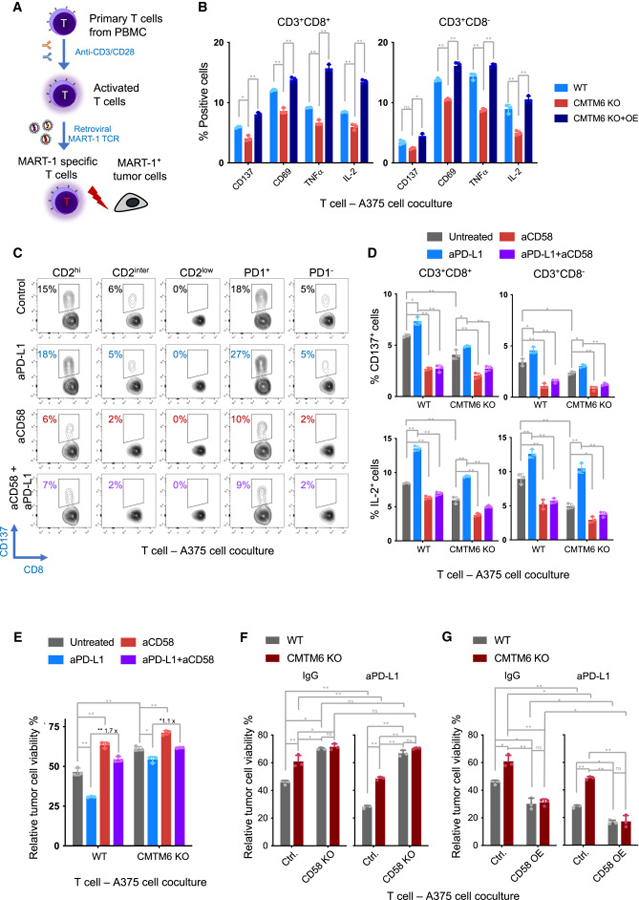
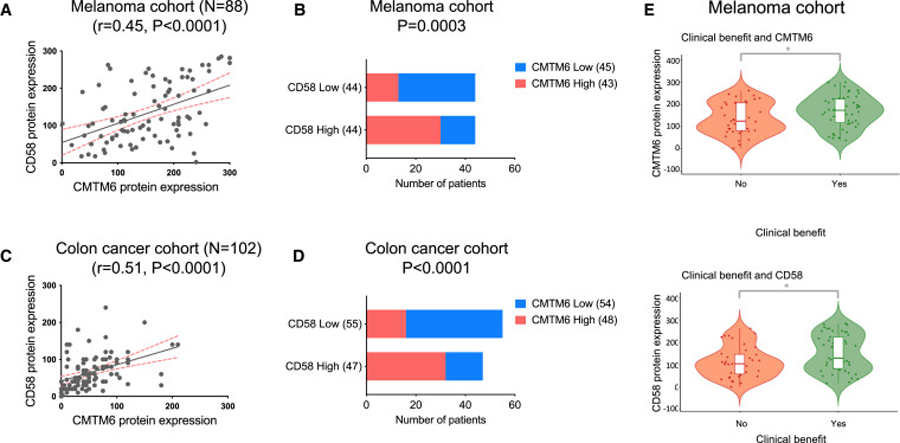
The dysregulated expression of immune checkpoint molecules enables cancer cells to evade immune destruction. While blockade of inhibitory immune checkpoints like PD-L1 forms the basis of current cancer immunotherapies, a deficiency in costimulatory signals can render these therapies futile. CD58, a costimulatory ligand, plays a crucial role in antitumor immune responses, but the mechanisms controlling its expression remain unclear. Using two systematic approaches, we reveal that CMTM6 positively regulates CD58 expression. Notably, CMTM6 interacts with both CD58 and PD-L1, maintaining the expression of these two immune checkpoint ligands with opposing functions. Functionally, the presence of CMTM6 and CD58 on tumor cells significantly affects T cell-tumor interactions and response to PD-L1-PD-1 blockade. Collectively, these findings provide fundamental insights into CD58 regulation, uncover a shared regulator of stimulatory and inhibitory immune checkpoints, and highlight the importance of tumor-intrinsic CMTM6 and CD58 expression in antitumor immune responses.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.M., T.R.B., T.N.M.S., and C.S. are listed as inventors on a patent application that covers the therapeutic and diagnostic use of CMTM6, CMTM4, and STUB1 as targets. C.S. has received research support from Bayer AG unrelated to the current study. T.N.M.S. serves as an advisor for Allogene Therapeutics, Celsius, Merus, Neogene Therapeutics, and Scenic Biotech, and is a stockholder in Allogene Therapeutics, Cell Control, Celsius, Merus, and Scenic Biotech. T.N.M.S. is also a venture partner at Third Rock Ventures, all outside the submitted work. T.R.B. is a cofounder and SAB member of Haplogen GmbH and cofounder and managing director of Scenic Biotech, all outside the submitted work. C.U.B. has received research grants from Novartis, BMS, and NanoString, is a paid advisory board member for BMS, MSD, Roche, Novartis, GlaxoSmithKline, AstraZeneca, Pfizer, Lilly, GenMab, and Pierre Fabre and holds ownership interest in Uniti Card, Neon Therapeutics, and Forty Seven, all outside this submitted work. S.W.L. is a founder and member of the scientific advisory board of Blueprint Medicines, Mirimus, ORIC Pharmaceuticals, and Faeth Therapeutics, and is on the scientific advisory board of Constellation Pharmaceuticals and PMV Pharmaceuticals, all outside the current work. H.M.H. reports consultancy fees from Roche Diagnostics paid to the institute and is an advisor for SlideScore and Ellogon from Roche Diagnostics, outside the submitted work. A.F. is currently employed at Bristol Myers Squibb, and R.G. is currently employed at Neogene Therapeutics; their contributions to this work were made prior to their respective positions and their current roles are not relevant to the content of this manuscript.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
