

Single-cell RNA sequencing captures patient-level heterogeneity and associated molecular phenotypes in breast cancer pleural effusions
- PMID: 37691350
- PMCID: PMC10493486
- DOI: 10.1002/ctm2.1356
Single-cell RNA sequencing captures patient-level heterogeneity and associated molecular phenotypes in breast cancer pleural effusions
Abstract
Background: Malignant pleural effusions (MPEs) are a common complication of advanced cancers, particularly those adjacent to the pleura, such as lung and breast cancer. The pathophysiology of MPE formation remains poorly understood, and although MPEs are routinely used for the diagnosis of breast cancer patients, their composition and biology are poorly understood. It is difficult to distinguish invading malignant cells from resident mesothelial cells and to identify the directionality of interactions between these populations in the pleura. There is a need to characterize the phenotypic diversity of breast cancer cell populations in the pleural microenvironment, and investigate how this varies across patients.
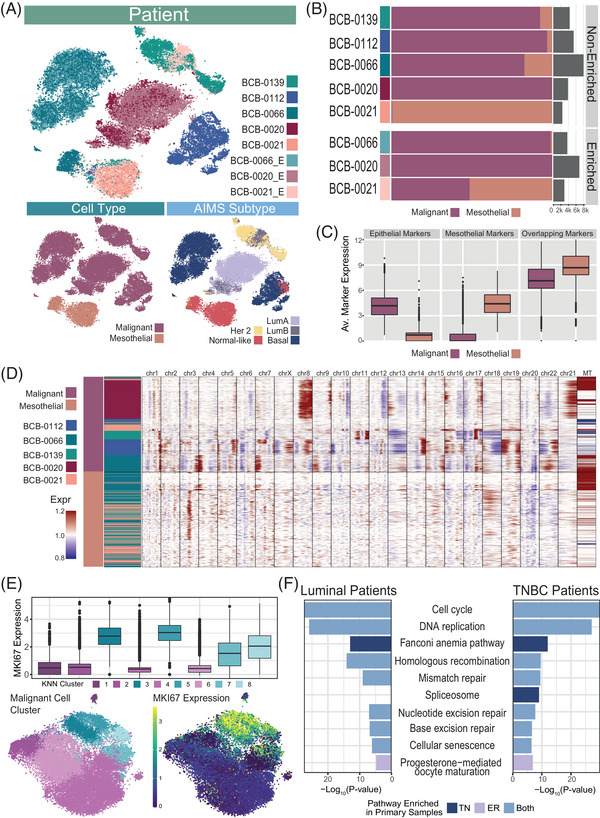
Methods: Here, we used single-cell RNA-sequencing to study the heterogeneity of 10 MPEs from seven metastatic breast cancer patients, including three Miltenyi-enriched samples using a negative selection approach. This dataset of almost 65 000 cells was analysed using integrative approaches to compare heterogeneous cell populations and phenotypes.
Results: We identified substantial inter-patient heterogeneity in the composition of cell types (including malignant, mesothelial and immune cell populations), in expression of subtype-specific gene signatures and in copy number aberration patterns, that captured variability across breast cancer cell populations. Within individual MPEs, we distinguished mesothelial cell populations from malignant cells using key markers, the presence of breast cancer subtype expression patterns and copy number aberration patterns. We also identified pleural mesothelial cells expressing a cancer-associated fibroblast-like transcriptomic program that may support cancer growth.
Conclusions: Our dataset presents the first unbiased assessment of breast cancer-associated MPEs at a single cell resolution, providing the community with a valuable resource for the study of MPEs. Our work highlights the molecular and cellular diversity captured in MPEs and motivates the potential use of these clinically relevant biopsies in the development of targeted therapeutics for patients with advanced breast cancer.
© 2023 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Malignant Pleural Effusion.2024 Aug 12. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Aug 12. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 34662055 Free Books & Documents.
-
Up-regulation of pro-angiogenic factors and establishment of tolerance in malignant pleural effusions.Lung Cancer. 2013 Oct;82(1):63-8. doi: 10.1016/j.lungcan.2013.07.007. Epub 2013 Aug 12. Lung Cancer. 2013. PMID: 23948549
-
Exploratory pilot study to characterize the immune landscapes of malignant pleural effusions and their corresponding primary tumors from patients with breast carcinoma and lung adenocarcinoma.J Am Soc Cytopathol. 2024 May-Jun;13(3):161-173. doi: 10.1016/j.jasc.2024.02.005. Epub 2024 Feb 29. J Am Soc Cytopathol. 2024. PMID: 38519275 Free PMC article.
-
Recent advances in the diagnosis and management of malignant pleural effusions.Mayo Clin Proc. 2008 Feb;83(2):235-50. doi: 10.4065/83.2.235. Mayo Clin Proc. 2008. PMID: 18241636 Review.
-
Molecular Profiling of Malignant Pleural Effusions with Next Generation Sequencing (NGS): Evidence that Supports Its Role in Cancer Management.J Pers Med. 2020 Nov 1;10(4):206. doi: 10.3390/jpm10040206. J Pers Med. 2020. PMID: 33139621 Free PMC article. Review.
Cited by
-
Missing cell types in single-cell references impact deconvolution of bulk data but are detectable.Genome Biol. 2025 Apr 7;26(1):86. doi: 10.1186/s13059-025-03506-9. Genome Biol. 2025. PMID: 40197327 Free PMC article.
-
VEGF Signal Complexity Confers Resistance to Atezolizumab, Bevacizumab, Carboplatin, and Paclitaxel in EGFR-Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer.MedComm (2020). 2025 Aug 19;6(9):e70335. doi: 10.1002/mco2.70335. eCollection 2025 Sep. MedComm (2020). 2025. PMID: 40843133 Free PMC article.
-
Intrapleural dual blockade of IL-6 and PD-L1 reprograms CAF dynamics and the tumor microenvironment in lung cancer-associated malignant pleural effusion.Respir Res. 2025 May 10;26(1):180. doi: 10.1186/s12931-025-03263-0. Respir Res. 2025. PMID: 40349069 Free PMC article.
-
Identification and characterization of tissue resident memory T cells in malignant pleural effusions associated with non-small cell lung cancer.Immunohorizons. 2025 Apr 26;9(6):vlaf013. doi: 10.1093/immhor/vlaf013. Immunohorizons. 2025. PMID: 40285480 Free PMC article.
References
-
- Antony VB, Loddenkemper R, Astoul P, et al. Management of malignant pleural effusions. Am J Respir Crit Care Med. 2000;162:1987‐2001. - PubMed
-
- Fentiman IS, Millis R, Sexton S, Hayward JL. Pleural effusion in breast cancer: a review of 105 cases. Cancer. 1981;47:2087‐2092. - PubMed
-
- Asciak R, Rahman NM. Malignant pleural effusion: from diagnostics to therapeutics. Clin Chest Med. 2018;39:181‐193. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases