

This is a preprint.
Computational exploration of the global microbiome for antibiotic discovery
- PMID: 37693522
- PMCID: PMC10491242
- DOI: 10.1101/2023.08.31.555663
Computational exploration of the global microbiome for antibiotic discovery
Update in
-
Discovery of antimicrobial peptides in the global microbiome with machine learning.Cell. 2024 Jul 11;187(14):3761-3778.e16. doi: 10.1016/j.cell.2024.05.013. Epub 2024 Jun 5. Cell. 2024. PMID: 38843834 Free PMC article.
Abstract
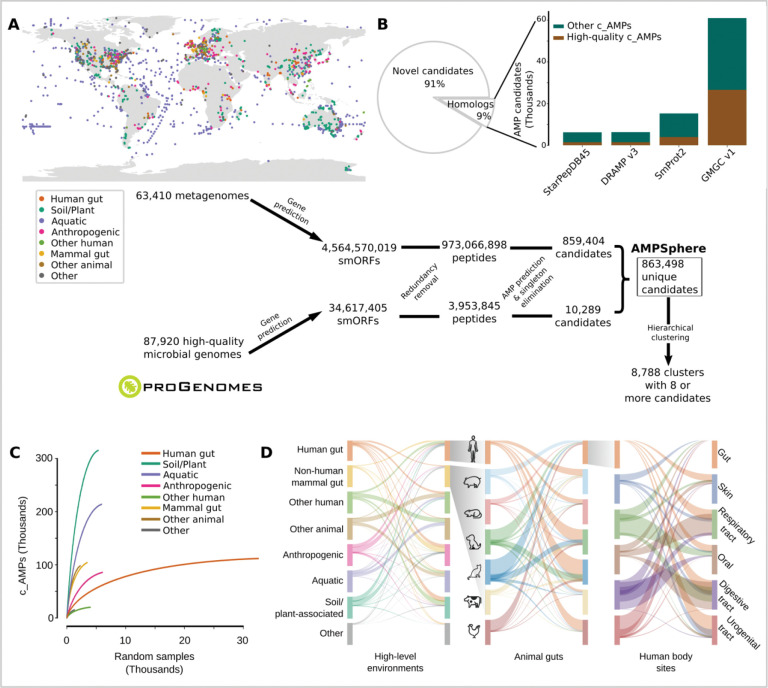
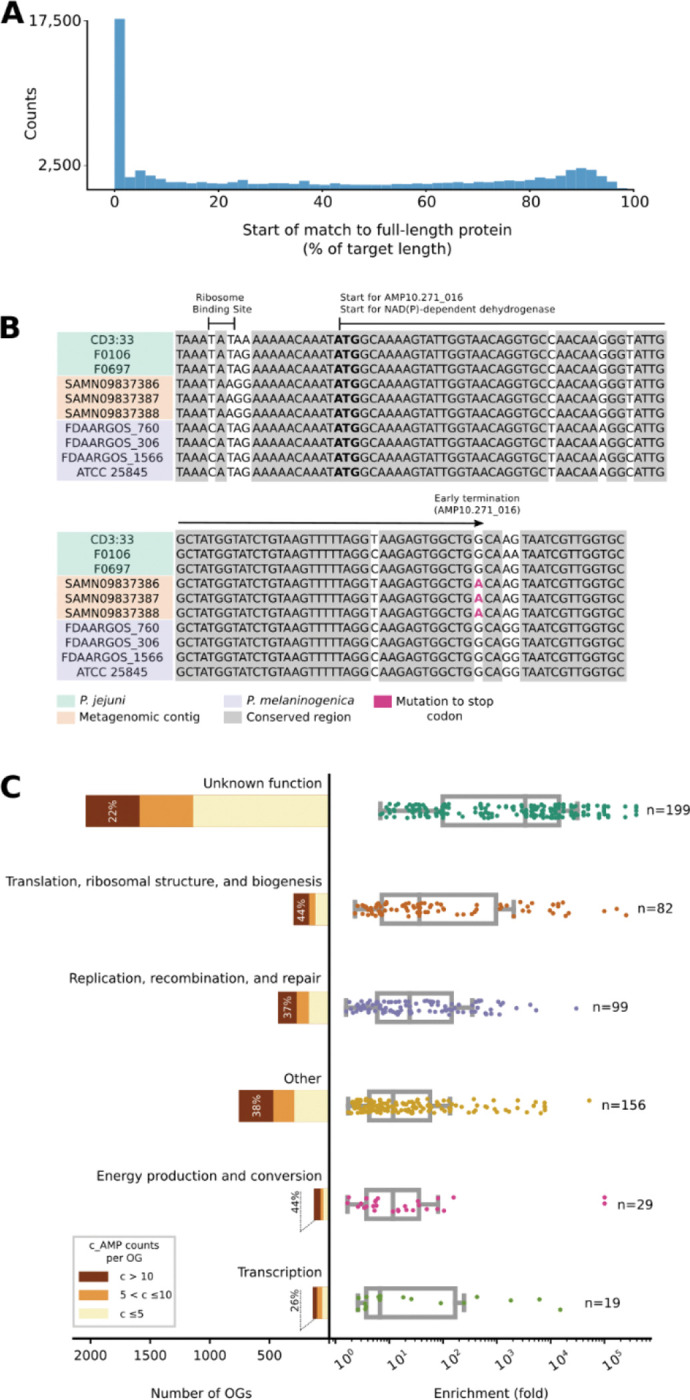
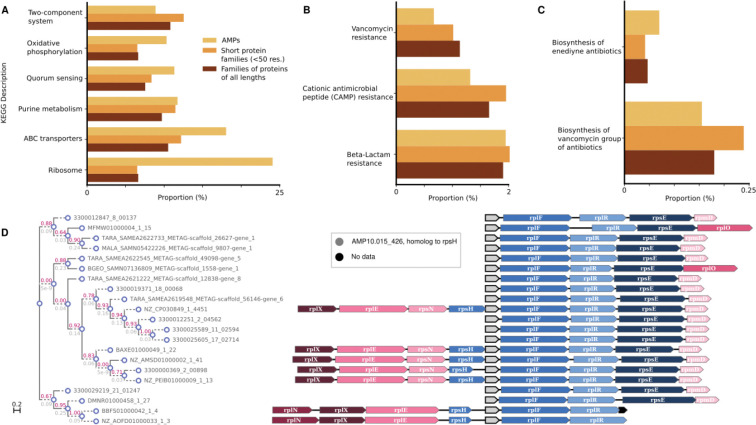
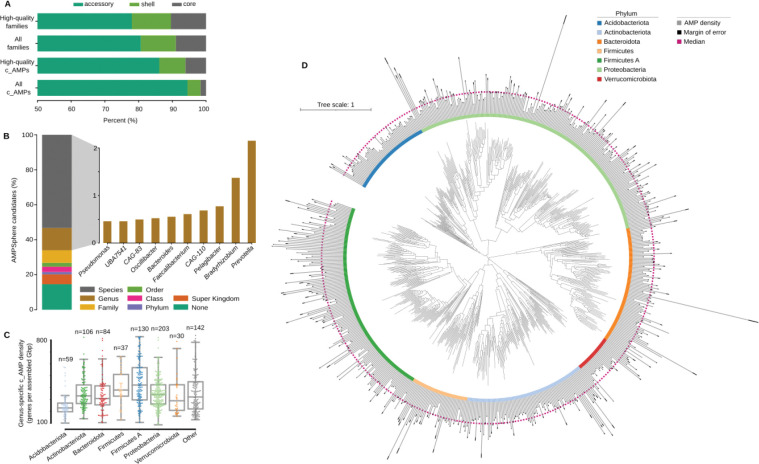
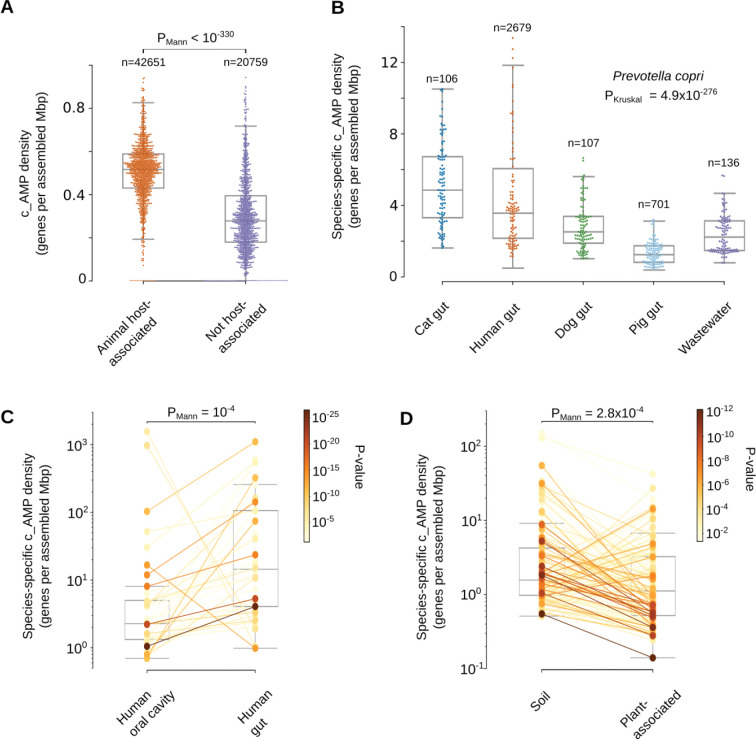
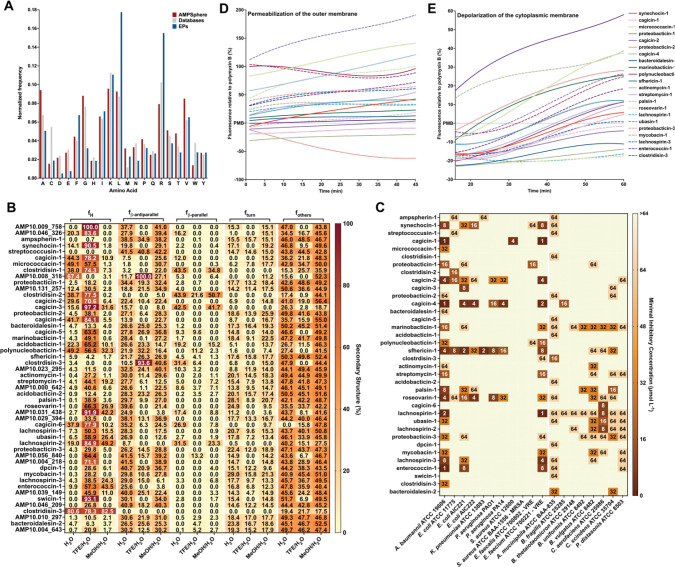
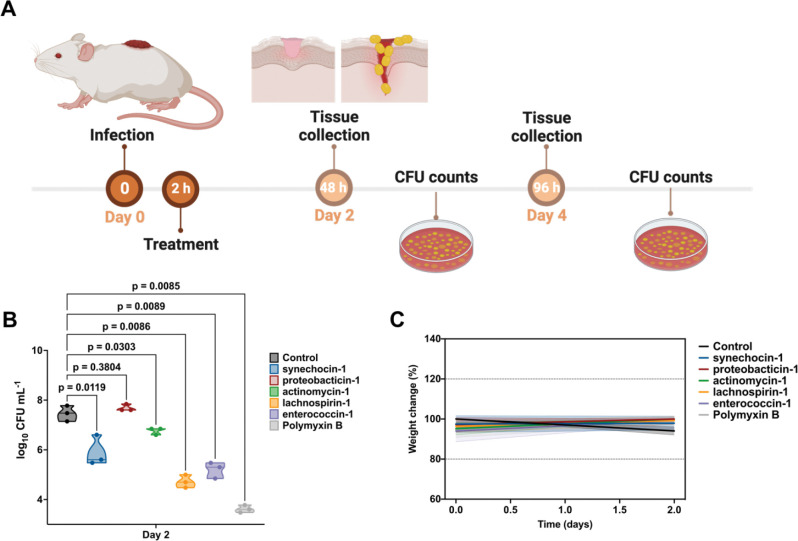
Novel antibiotics are urgently needed to combat the antibiotic-resistance crisis. We present a machine learning-based approach to predict prokaryotic antimicrobial peptides (AMPs) by leveraging a vast dataset of 63,410 metagenomes and 87,920 microbial genomes. This led to the creation of AMPSphere, a comprehensive catalog comprising 863,498 non-redundant peptides, the majority of which were previously unknown. We observed that AMP production varies by habitat, with animal-associated samples displaying the highest proportion of AMPs compared to other habitats. Furthermore, within different human-associated microbiota, strain-level differences were evident. To validate our predictions, we synthesized and experimentally tested 50 AMPs, demonstrating their efficacy against clinically relevant drug-resistant pathogens both in vitro and in vivo. These AMPs exhibited antibacterial activity by targeting the bacterial membrane. Additionally, AMPSphere provides valuable insights into the evolutionary origins of peptides. In conclusion, our approach identified AMP sequences within prokaryotic microbiomes, opening up new avenues for the discovery of antibiotics.
Keywords: antimicrobial activity; antimicrobial peptides; global microbiome; machine learning; metagenomics.
Conflict of interest statement
Declaration of interests Cesar de la Fuente-Nunez provides consulting services to Invaio Sciences and is a member of the Scientific Advisory Boards of Nowture S.L. and Phare Bio. The de la Fuente Lab has received research funding or in-kind donations from United Therapeutics, Strata Manufacturing PJSC, and Procter & Gamble, none of which were used in support of this work. All other authors state they do not have any competing interests.
Figures
References
-
- Porto W.F., Irazazabal L., Alves E.S.F., Ribeiro S.M., Matos C.O., Pires Á.S., Fensterseifer I.C.M., Miranda V.J., Haney E.F., Humblot V., et al. (2018). In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat Commun 9, 1490. 10.1038/s41467-018-03746-3. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources