

Nucleosome density shapes kilobase-scale regulation by a mammalian chromatin remodeler
- PMID: 37696956
- PMCID: PMC10584690
- DOI: 10.1038/s41594-023-01093-6
Nucleosome density shapes kilobase-scale regulation by a mammalian chromatin remodeler
Abstract
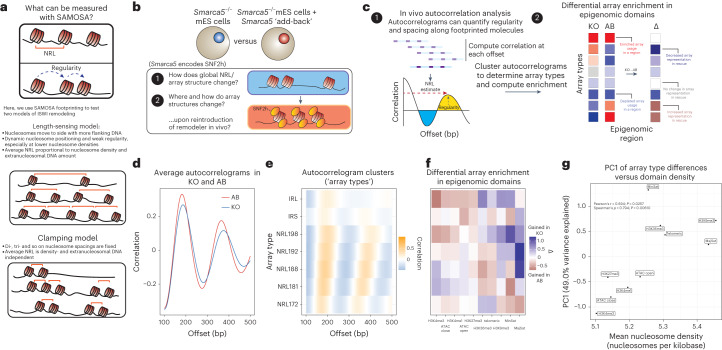
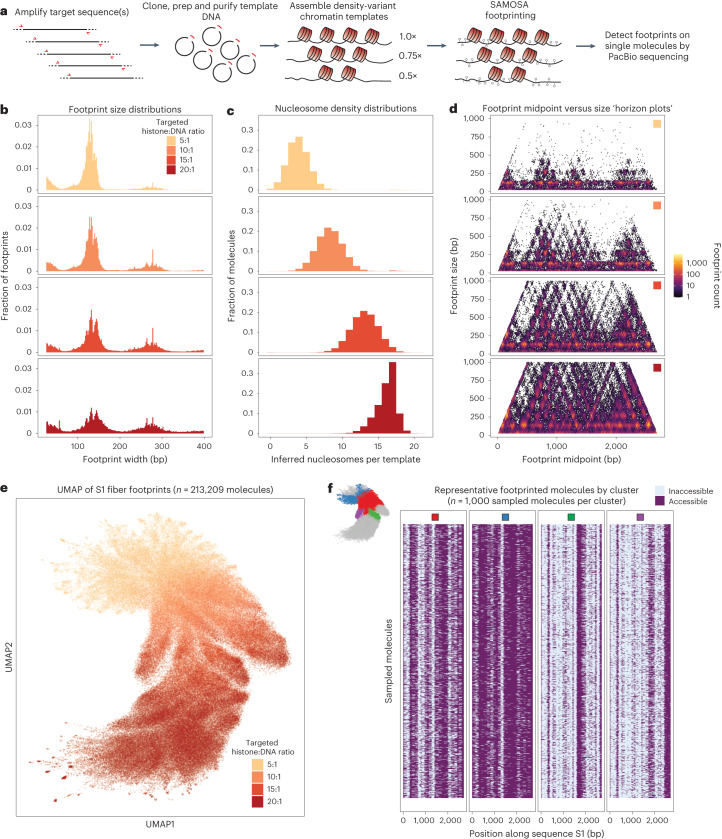
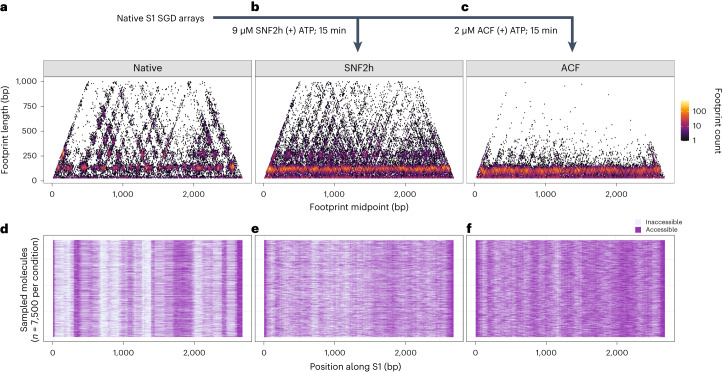
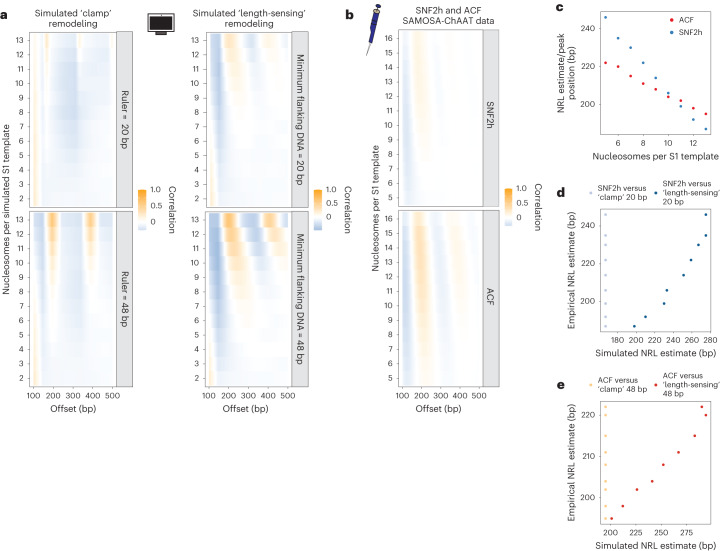
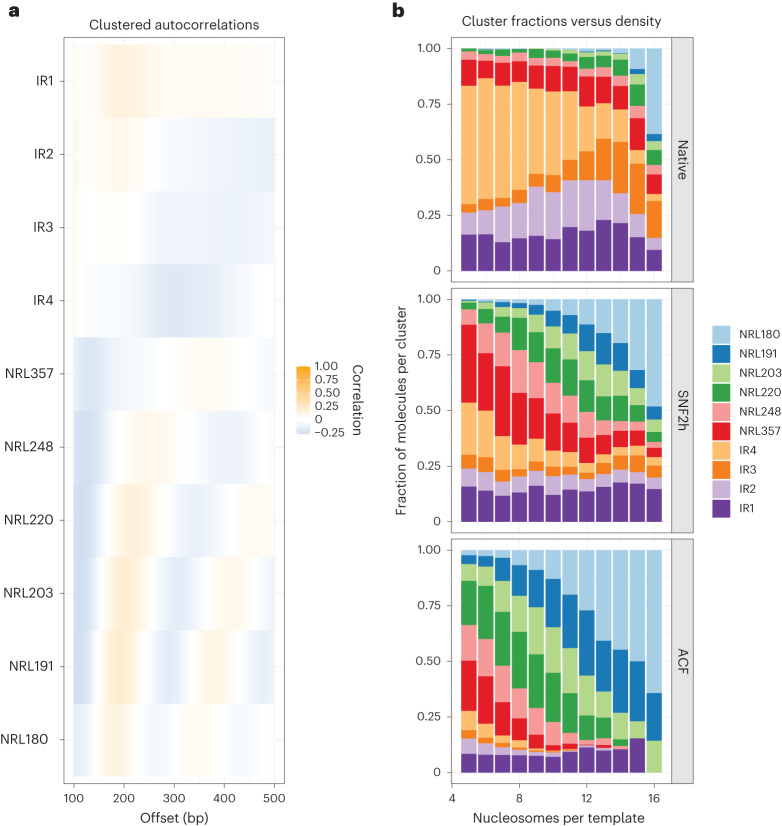
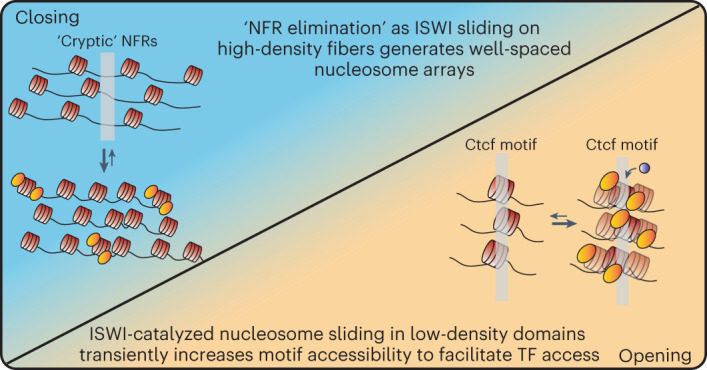
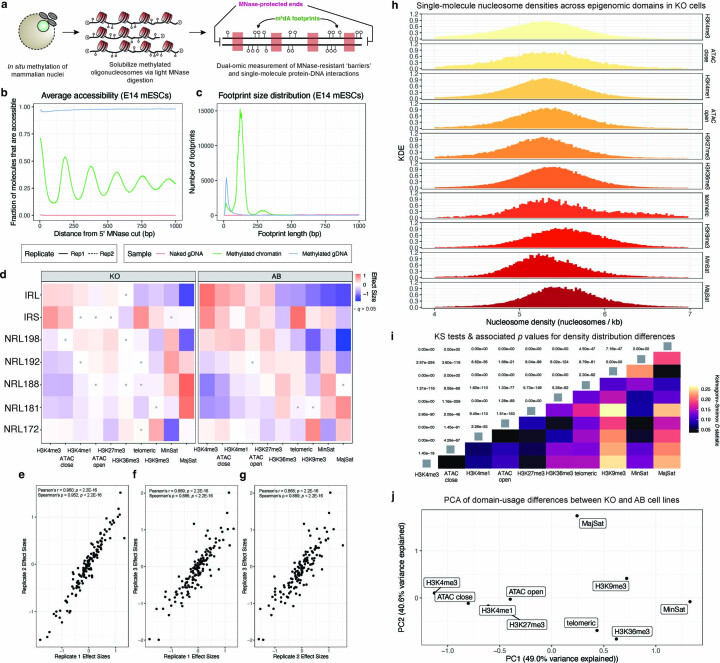
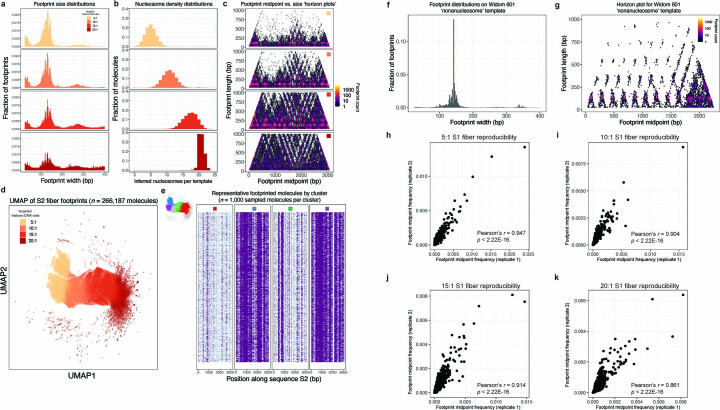
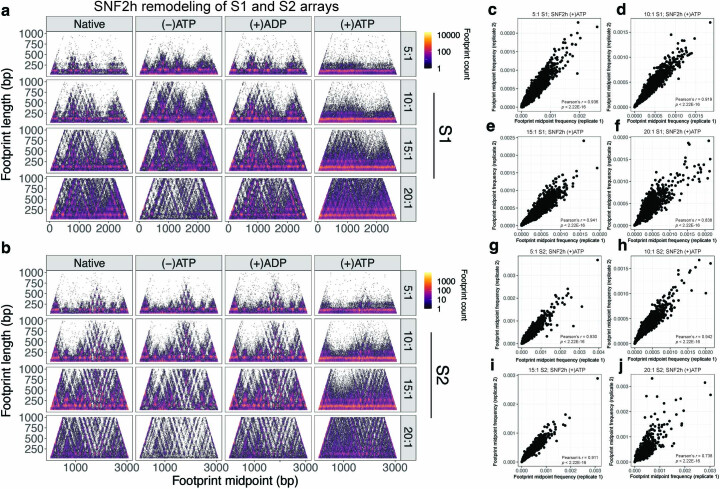
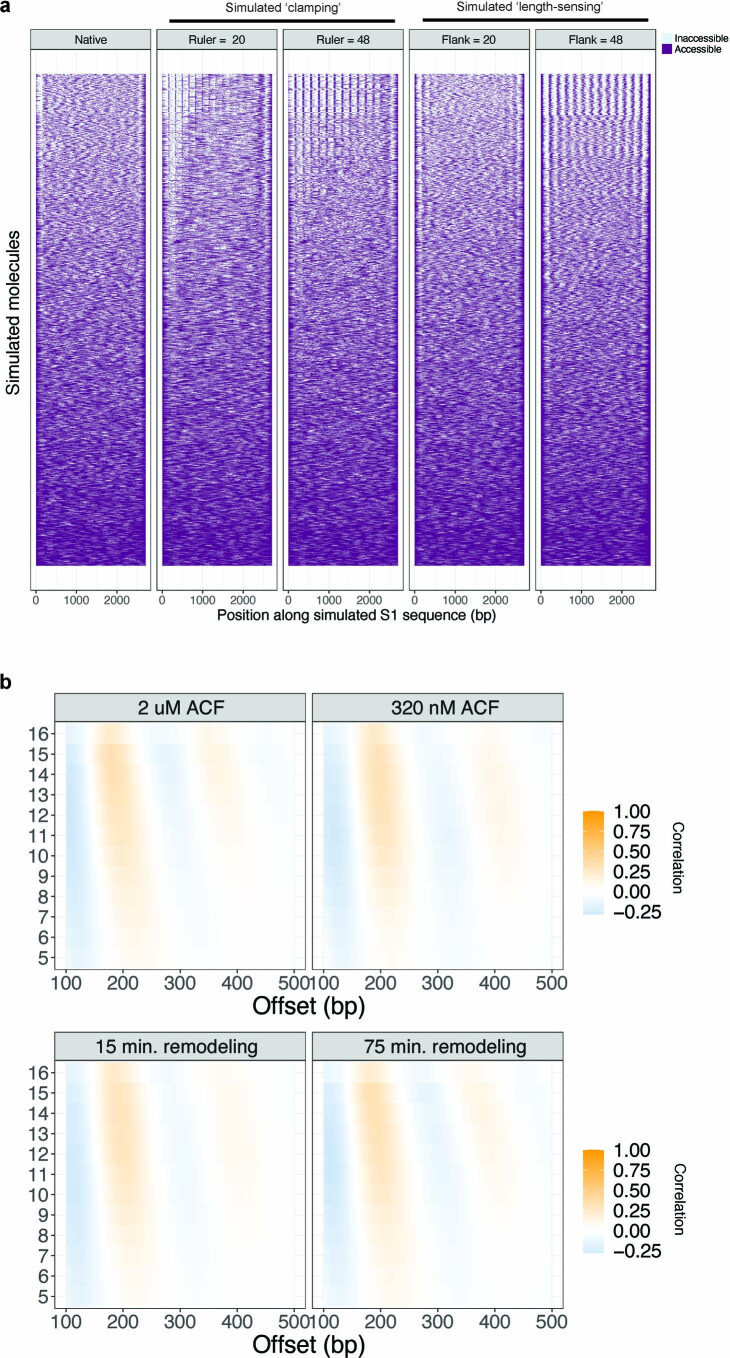
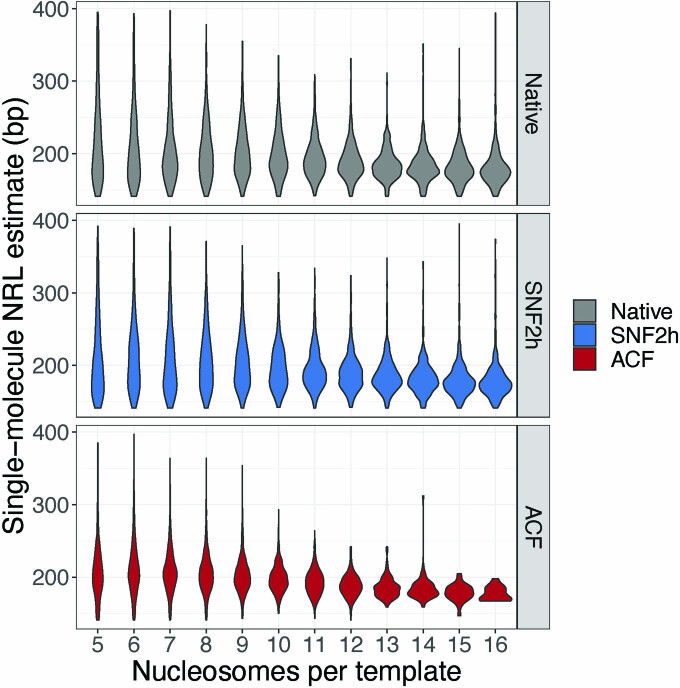
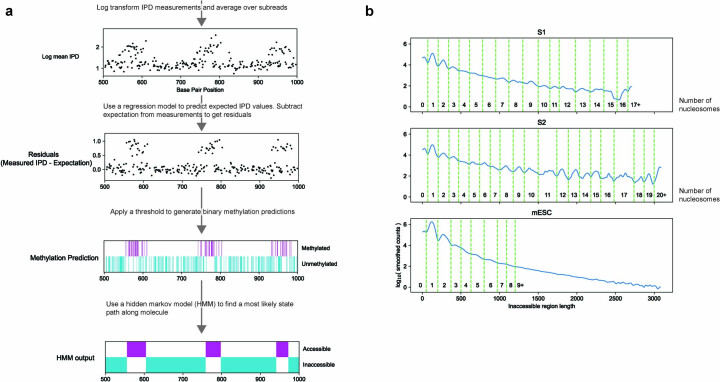
Nearly all essential nuclear processes act on DNA packaged into arrays of nucleosomes. However, our understanding of how these processes (for example, DNA replication, RNA transcription, chromatin extrusion and nucleosome remodeling) occur on individual chromatin arrays remains unresolved. Here, to address this deficit, we present SAMOSA-ChAAT: a massively multiplex single-molecule footprinting approach to map the primary structure of individual, reconstituted chromatin templates subject to virtually any chromatin-associated reaction. We apply this method to distinguish between competing models for chromatin remodeling by the essential imitation switch (ISWI) ATPase SNF2h: nucleosome-density-dependent spacing versus fixed-linker-length nucleosome clamping. First, we perform in vivo single-molecule nucleosome footprinting in murine embryonic stem cells, to discover that ISWI-catalyzed nucleosome spacing correlates with the underlying nucleosome density of specific epigenomic domains. To establish causality, we apply SAMOSA-ChAAT to quantify the activities of ISWI ATPase SNF2h and its parent complex ACF on reconstituted nucleosomal arrays of varying nucleosome density, at single-molecule resolution. We demonstrate that ISWI remodelers operate as density-dependent, length-sensing nucleosome sliders, whose ability to program DNA accessibility is dictated by single-molecule nucleosome density. We propose that the long-observed, context-specific regulatory effects of ISWI complexes can be explained in part by the sensing of nucleosome density within epigenomic domains. More generally, our approach promises molecule-precise views of the essential processes that shape nuclear physiology.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
