

Rinmaker: a fast, versatile and reliable tool to determine residue interaction networks in proteins
- PMID: 37697267
- PMCID: PMC10496328
- DOI: 10.1186/s12859-023-05466-y
Rinmaker: a fast, versatile and reliable tool to determine residue interaction networks in proteins
Abstract
Background: Residue Interaction Networks (RINs) map the crystallographic description of a protein into a graph, where amino acids are represented as nodes and non-covalent bonds as edges. Determination and visualization of a protein as a RIN provides insights on the topological properties (and hence their related biological functions) of large proteins without dealing with the full complexity of the three-dimensional description, and hence it represents an invaluable tool of modern bioinformatics.
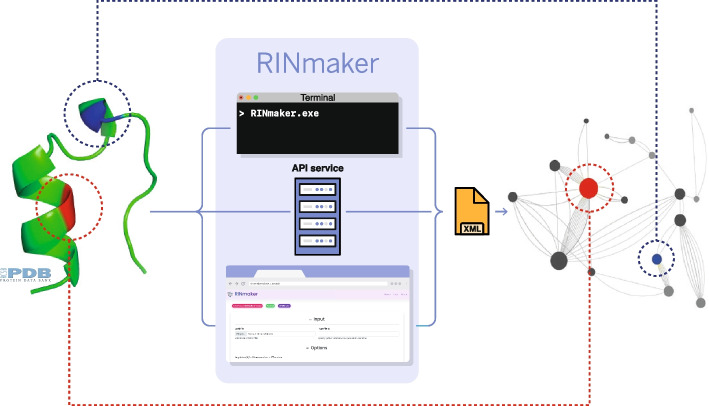
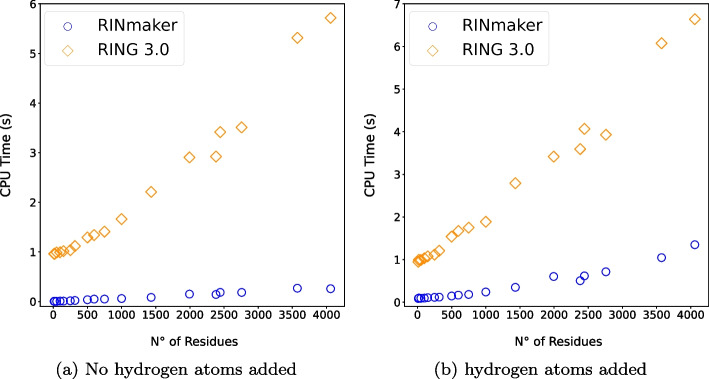
Results: We present RINmaker, a fast, flexible, and powerful tool for determining and visualizing RINs that include all standard non-covalent interactions. RINmaker is offered as a cross-platform and open source software that can be used either as a command-line tool or through a web application or a web API service. We benchmark its efficiency against the main alternatives and provide explicit tests to show its performance and its correctness.
Conclusions: RINmaker is designed to be fully customizable, from a simple and handy support for experimental research to a sophisticated computational tool that can be embedded into a large computational pipeline. Hence, it paves the way to bridge the gap between data-driven/machine learning approaches and numerical simulations of simple, physically motivated, models.
Keywords: Non-covalent bonds; Protein 3D structure; Residue interaction network (RIN).
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
