

Tracking reactive astrogliosis in autosomal dominant and sporadic Alzheimer's disease with multi-modal PET and plasma GFAP
- PMID: 37697307
- PMCID: PMC10496408
- DOI: 10.1186/s13024-023-00647-y
Tracking reactive astrogliosis in autosomal dominant and sporadic Alzheimer's disease with multi-modal PET and plasma GFAP
Abstract
Background: Plasma assays for the detection of Alzheimer's disease neuropathological changes are receiving ever increasing interest. The concentration of plasma glial fibrillary acidic protein (GFAP) has been suggested as a potential marker of astrocytes or recently, amyloid-β burden, although this hypothesis remains unproven. We compared plasma GFAP levels with the astrocyte tracer 11C-Deuterium-L-Deprenyl (11C-DED) in a multi-modal PET design in participants with sporadic and Autosomal Dominant Alzheimer's disease.
Methods: Twenty-four individuals from families with known Autosomal Dominant Alzheimer's Disease mutations (mutation carriers = 10; non-carriers = 14) and fifteen patients with sporadic Alzheimer's disease were included. The individuals underwent PET imaging with 11C-DED, 11C-PIB and 18F-FDG, as markers of reactive astrogliosis, amyloid-β deposition, and glucose metabolism, respectively, and plasma sampling for measuring GFAP concentrations. Twenty-one participants from the Autosomal Dominant Alzheimer's Disease group underwent follow-up plasma sampling and ten of these participants underwent follow-up PET imaging.
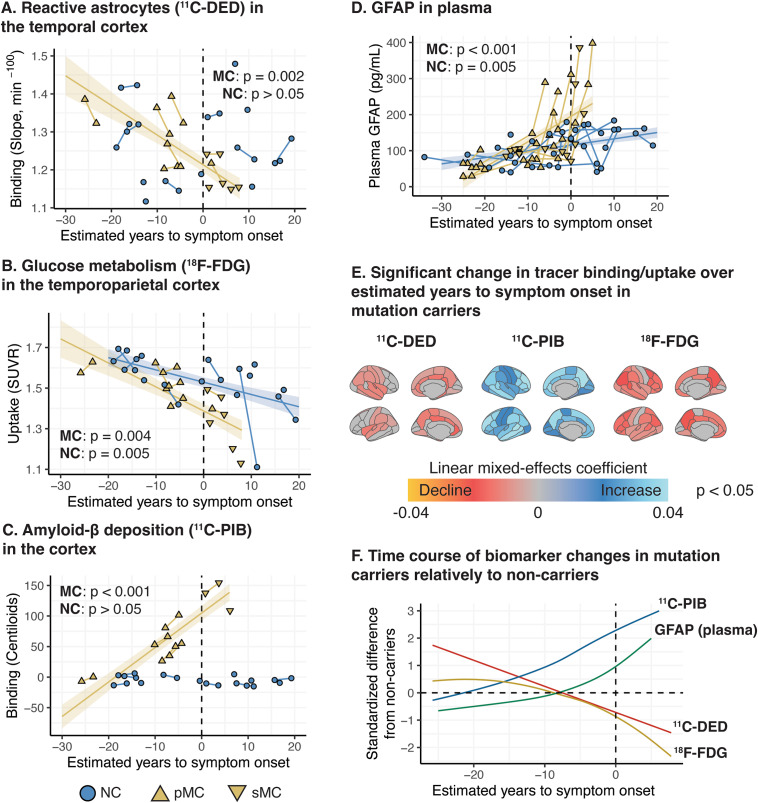
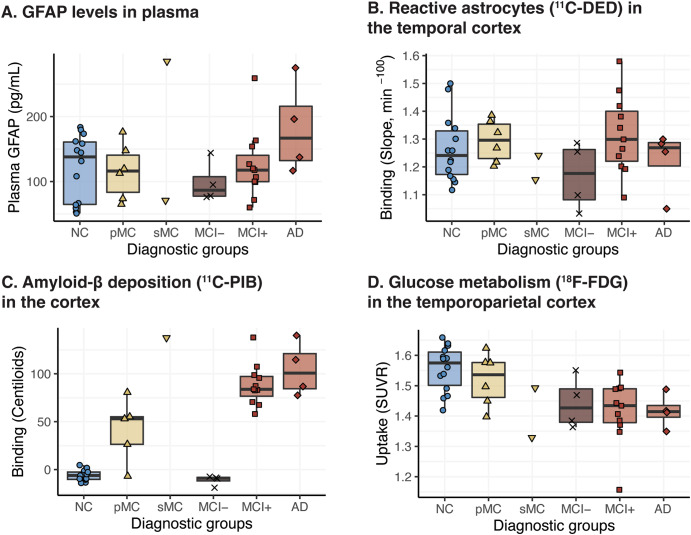
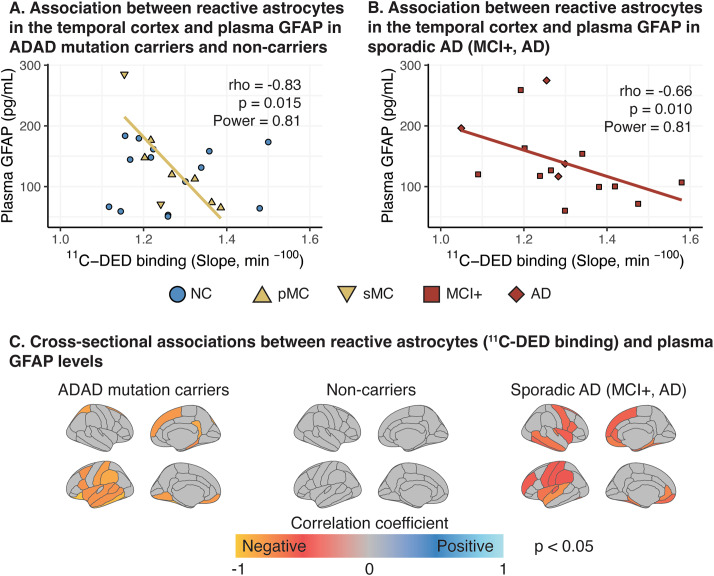
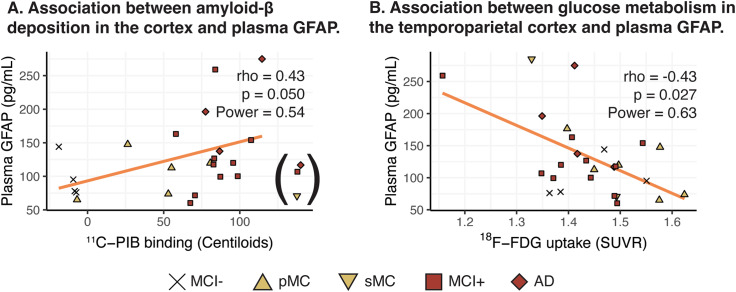
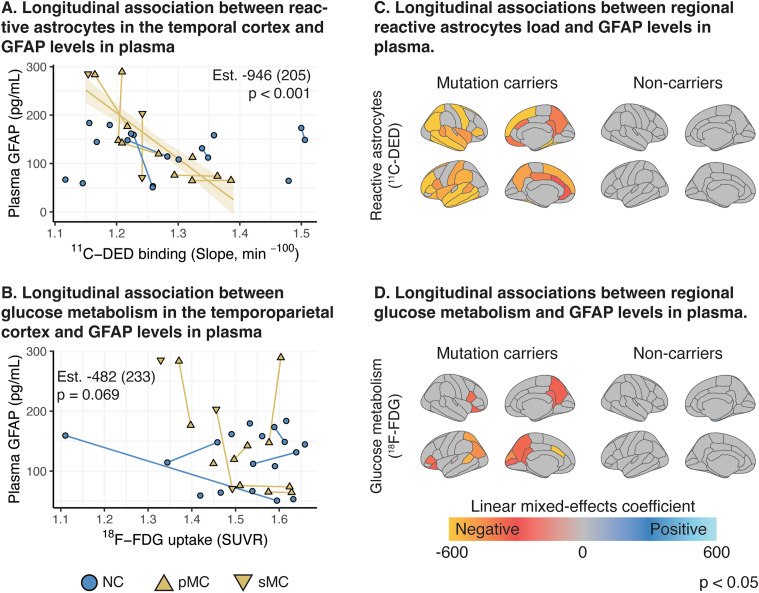
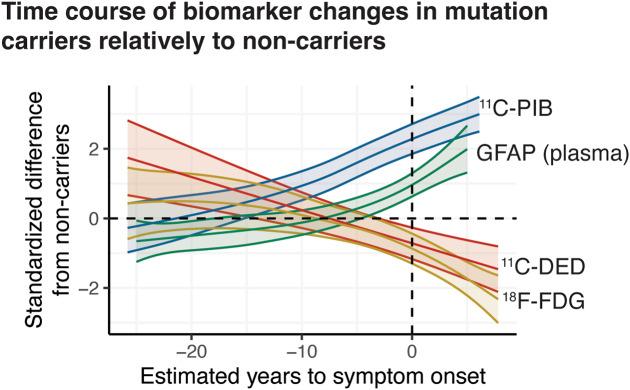
Results: In mutation carriers, plasma GFAP levels and 11C-PIB binding increased, while 11C-DED binding and 18F-FDG uptake significantly decreased across the estimated years to symptom onset. Cross-sectionally, plasma GFAP demonstrated a negative correlation with 11C-DED binding in both mutation carriers and patients with sporadic disease. Plasma GFAP indicated cross-sectionally a significant positive correlation with 11C-PIB binding and a significant negative correlation with 18F-FDG in the whole sample. The longitudinal levels of 11C-DED binding showed a significant negative correlation with longitudinal plasma GFAP concentrations over the follow-up interval.
Conclusions: Plasma GFAP concentration and astrocyte 11C-DED brain binding levels followed divergent trajectories and may reflect different underlying processes. The strong negative association between plasma GFAP and 11C-DED binding in Autosomal Dominant and sporadic Alzheimer's disease brains may indicate that if both are markers of reactive astrogliosis, they may detect different states or subtypes of astrogliosis. Increased 11C-DED brain binding seems to be an earlier phenomenon in Alzheimer's disease progression than increased plasma GFAP concentration.
Keywords: Alzheimer’s disease; Astrocytes; Astrogliosis; Autosomal Dominant Alzheimer’s disease; Deprenyl; Plasma GFAP.
© 2023. Editorial Group and BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
KC, CJ, and ERV have no competing interests. AN has received a consulting fee from AVVA Pharmaceuticals, H Lundbeck A/S, Hoffman La Roche, honorarium for lecture Hoffman La Roche, Roche and hold a patent WO 2022/255915. Patent No. PCT/SE2022/050413 PET Tracers. HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work).
Figures
References
-
- Kumar A, Fontana IC, Nordberg A. Reactive astrogliosis: a friend or foe in the pathogenesis of Alzheimer’s disease. J Neurochem. 2022;jnc.15565. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
