

PhaseDancer: a novel targeted assembler of segmental duplications unravels the complexity of the human chromosome 2 fusion going from 48 to 46 chromosomes in hominin evolution
- PMID: 37697406
- PMCID: PMC10496407
- DOI: 10.1186/s13059-023-03022-8
PhaseDancer: a novel targeted assembler of segmental duplications unravels the complexity of the human chromosome 2 fusion going from 48 to 46 chromosomes in hominin evolution
Abstract
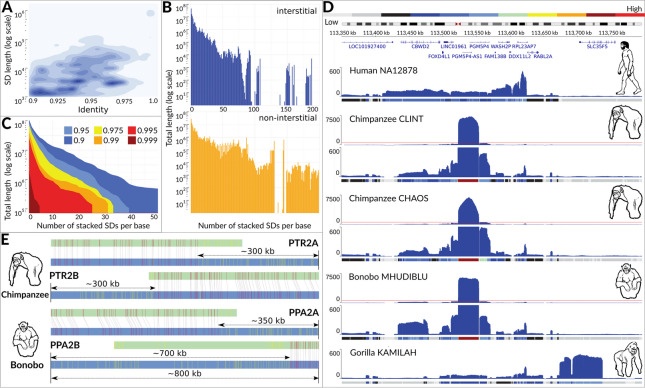
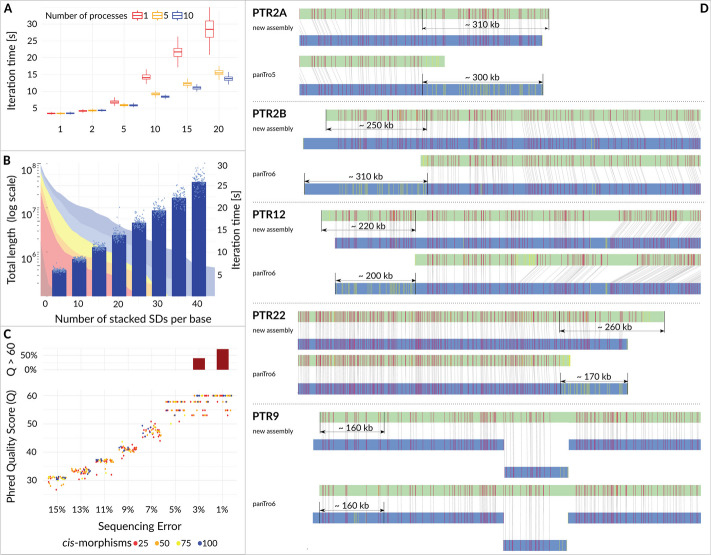
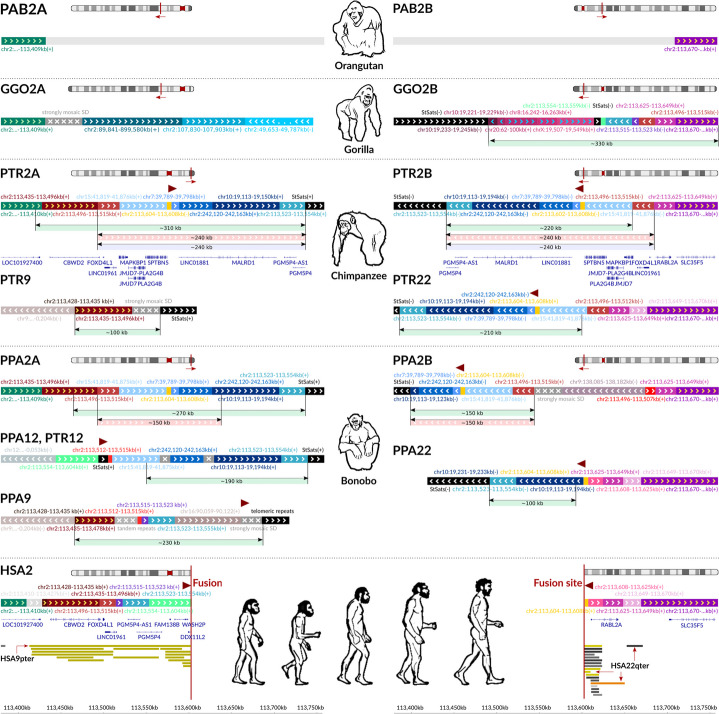
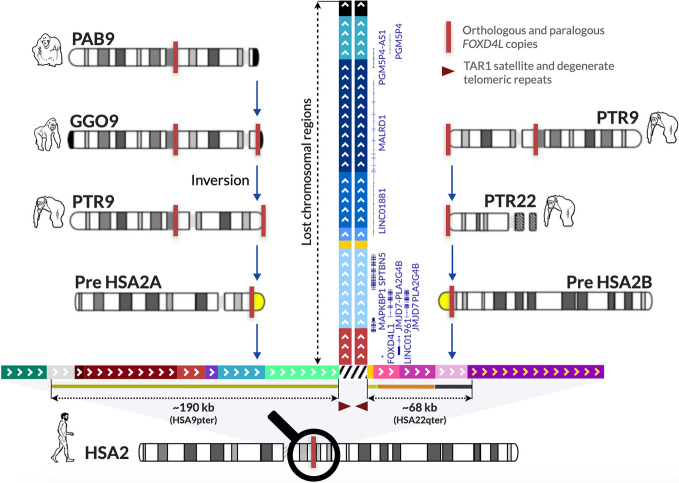
Resolving complex genomic regions rich in segmental duplications (SDs) is challenging due to the high error rate of long-read sequencing. Here, we describe a targeted approach with a novel genome assembler PhaseDancer that extends SD-rich regions of interest iteratively. We validate its robustness and efficiency using a golden-standard set of human BAC clones and in silico-generated SDs with predefined evolutionary scenarios. PhaseDancer enables extension of the incomplete complex SD-rich subtelomeric regions of Great Ape chromosomes orthologous to the human chromosome 2 (HSA2) fusion site, informing a model of HSA2 formation and unravelling the evolution of human and Great Ape genomes.
Keywords: Chromosomal fusion; Complex genomic rearrangements; De-novo assembly; Long-read PacBio sequencing; Segmental duplications.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Sohn J-I, Nam J-W. The present and future of de novo whole-genome assembly. Brief Bioinforma. 2016;096. 10.1093/bib/bbw096. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
