

Altered Smooth Muscle Cell Histone Acetylome by the SPHK2/S1P Axis Promotes Pulmonary Hypertension
- PMID: 37698017
- PMCID: PMC10543610
- DOI: 10.1161/CIRCRESAHA.123.322740
Altered Smooth Muscle Cell Histone Acetylome by the SPHK2/S1P Axis Promotes Pulmonary Hypertension
Abstract
Background: Epigenetic regulation of vascular remodeling in pulmonary hypertension (PH) is poorly understood. Transcription regulating, histone acetylation code alters chromatin accessibility to promote transcriptional activation. Our goal was to identify upstream mechanisms that disrupt epigenetic equilibrium in PH.
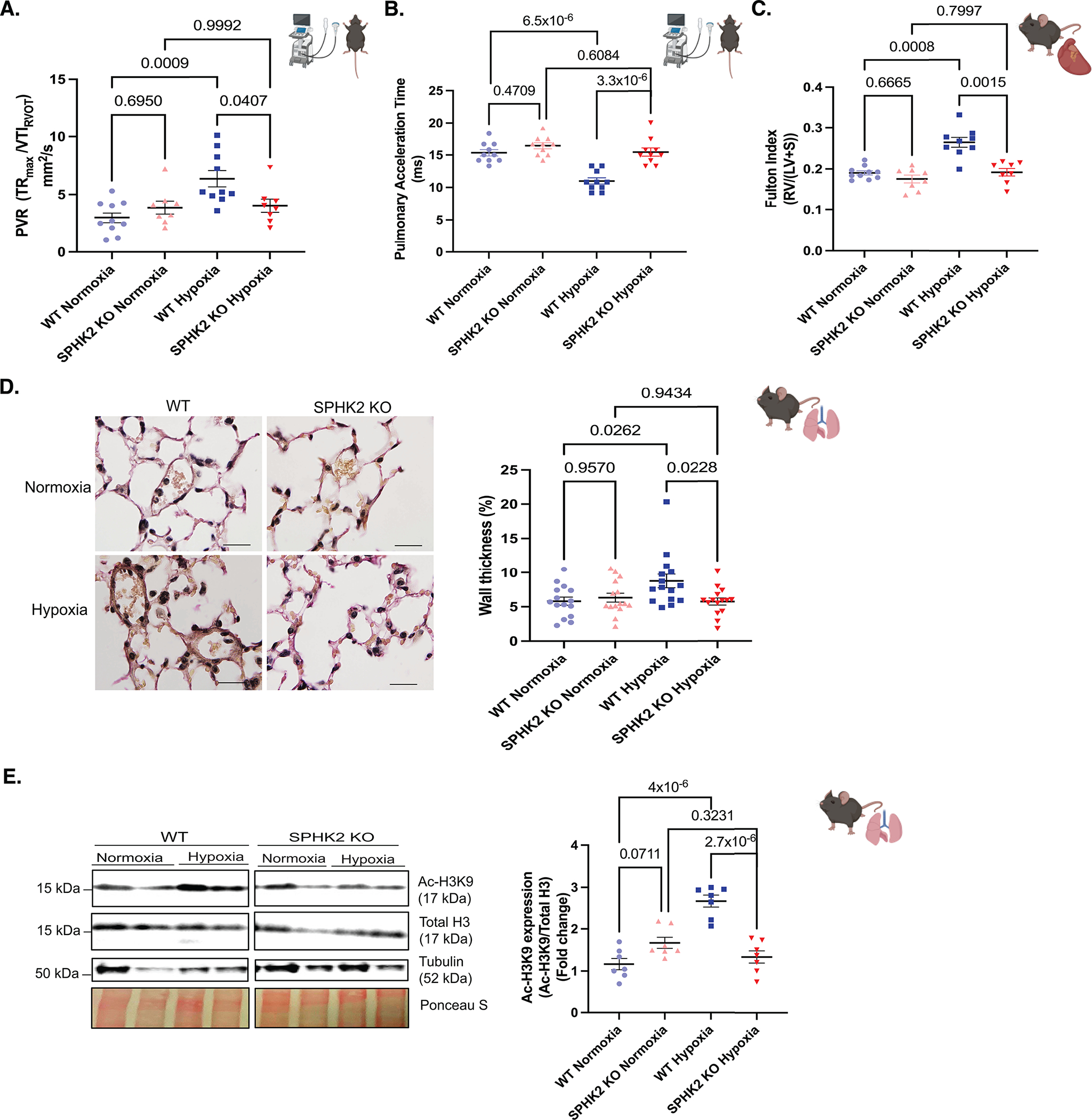
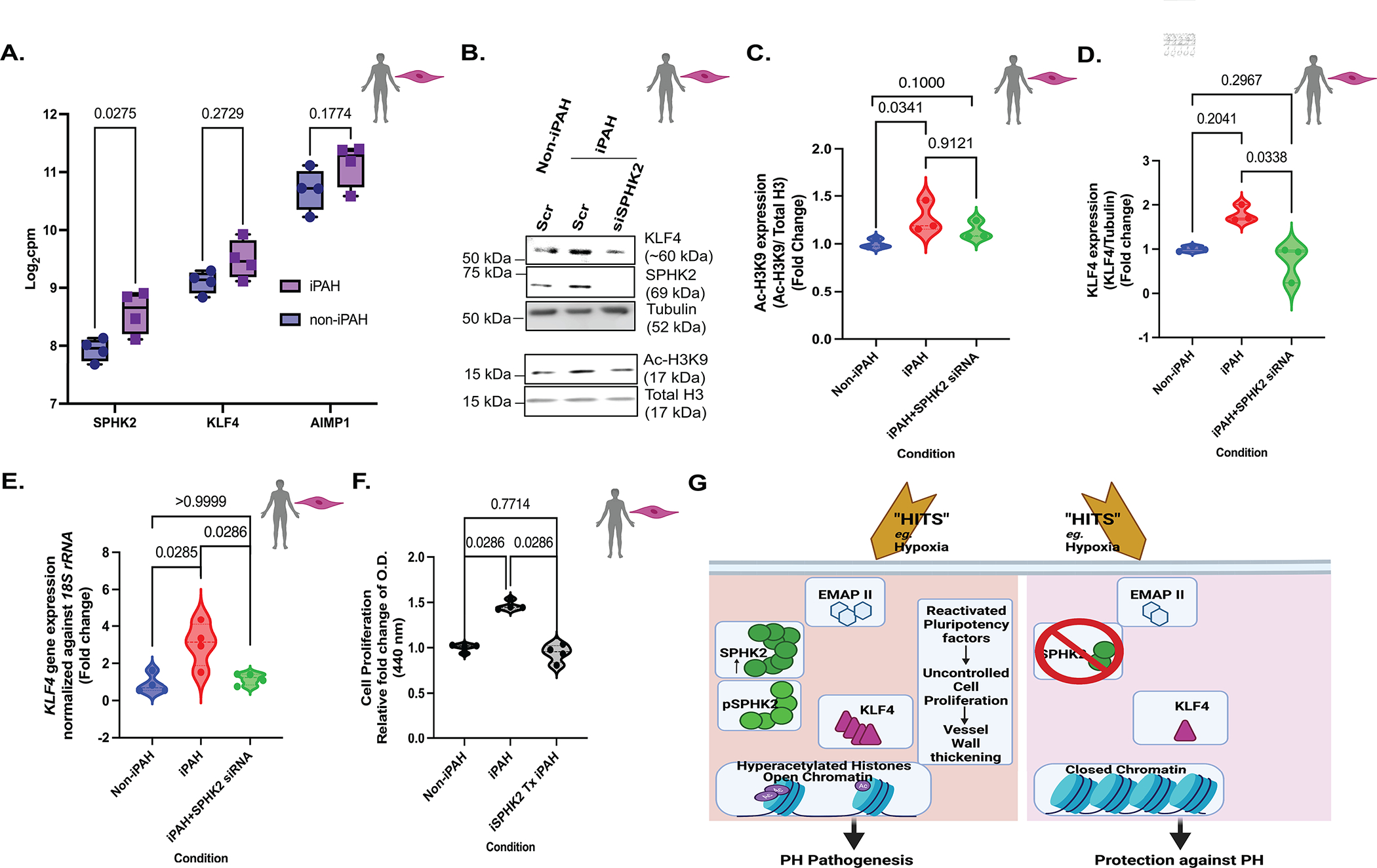
Methods: Human pulmonary artery smooth muscle cells (PASMCs), human idiopathic pulmonary arterial hypertension (iPAH):human PASMCs, iPAH lung tissue, failed donor lung tissue, human pulmonary microvascular endothelial cells, iPAH:PASMC and non-iPAH:PASMC RNA-seq databases, NanoString nCounter, and cleavage under targets and release using nuclease were utilized to investigate histone acetylation, hyperacetylation targets, protein and gene expression, sphingolipid activation, cell proliferation, and gene target identification. SPHK2 (sphingosine kinase 2) knockout was compared with control C57BL/6NJ mice after 3 weeks of hypoxia and assessed for indices of PH.
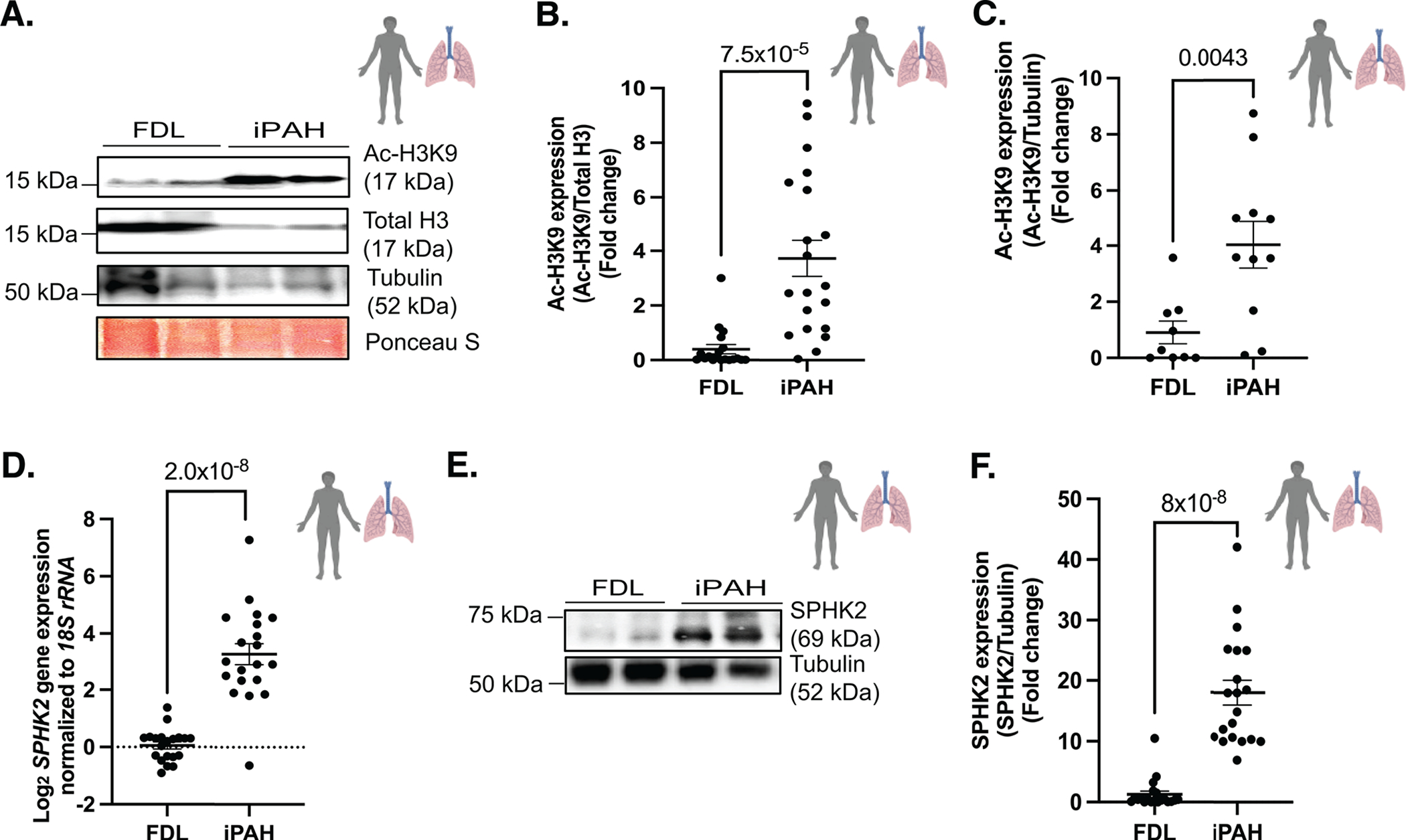
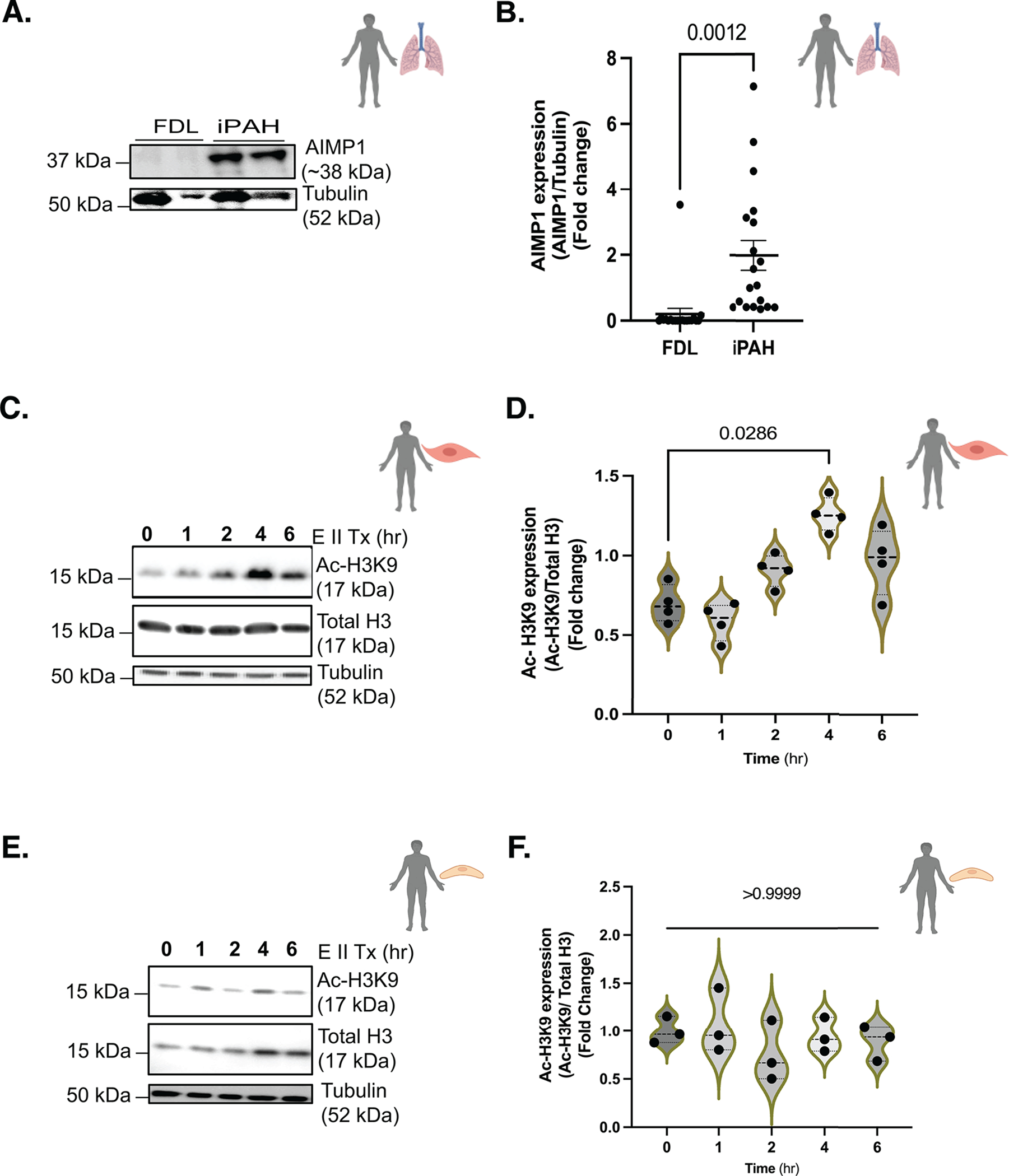
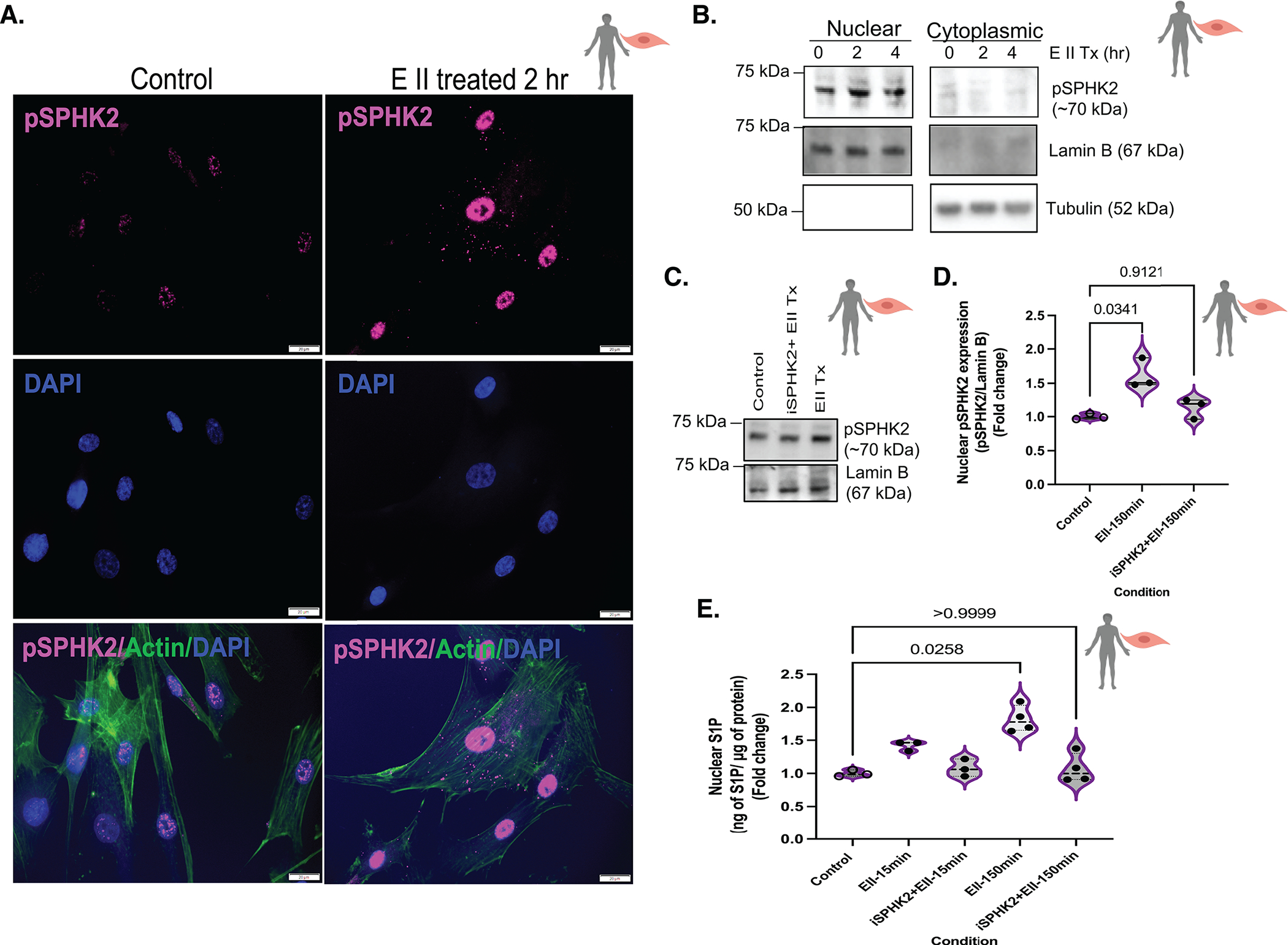
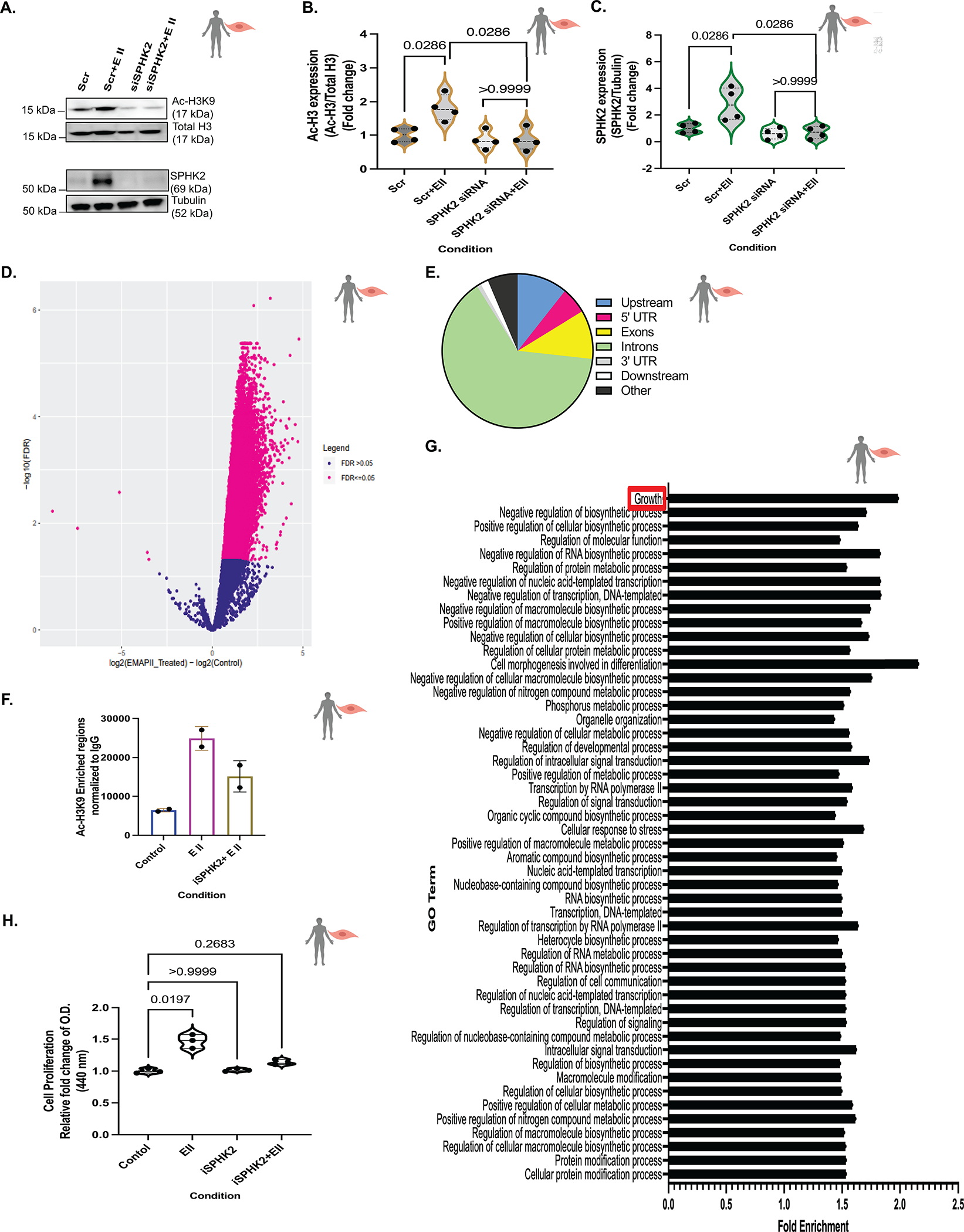
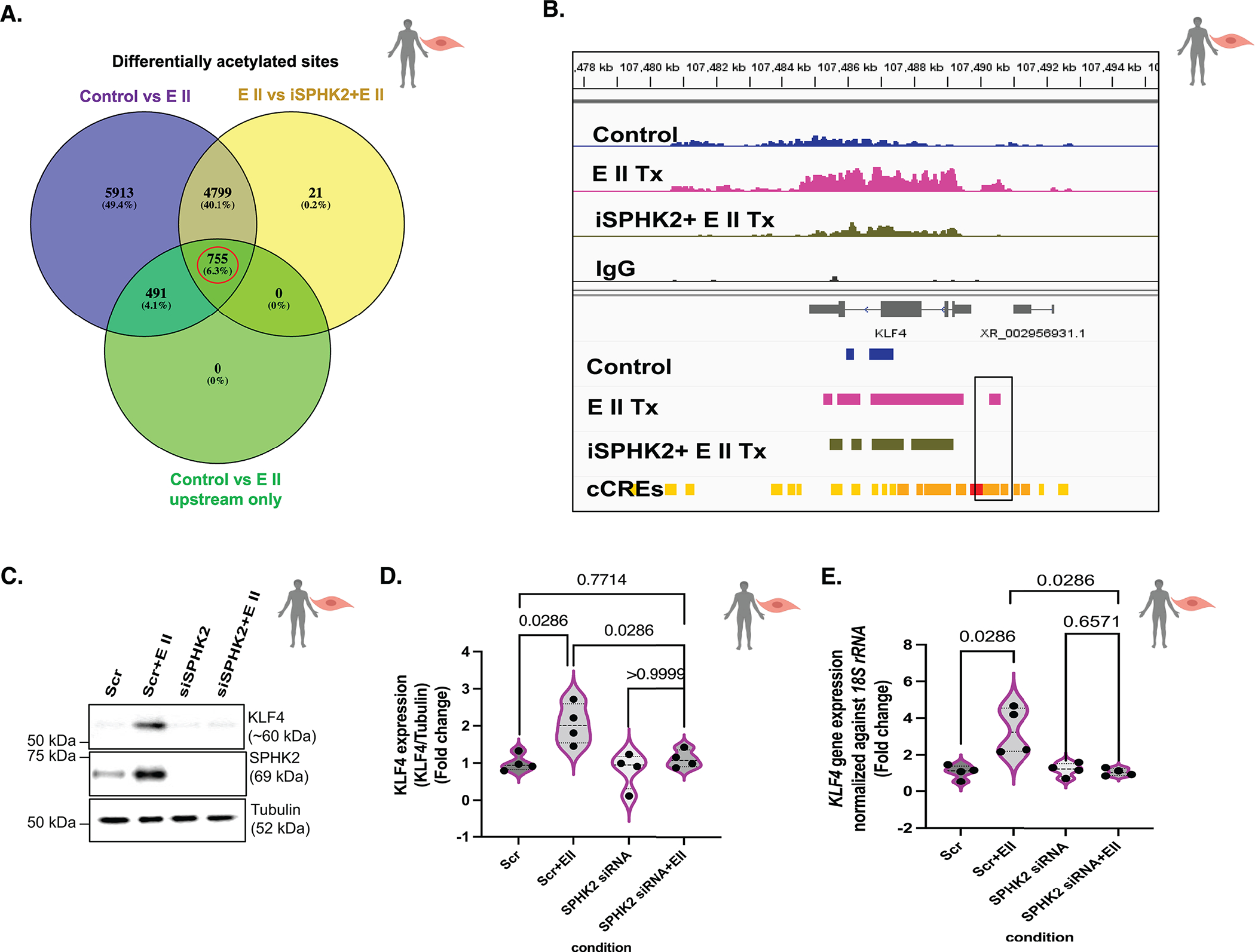
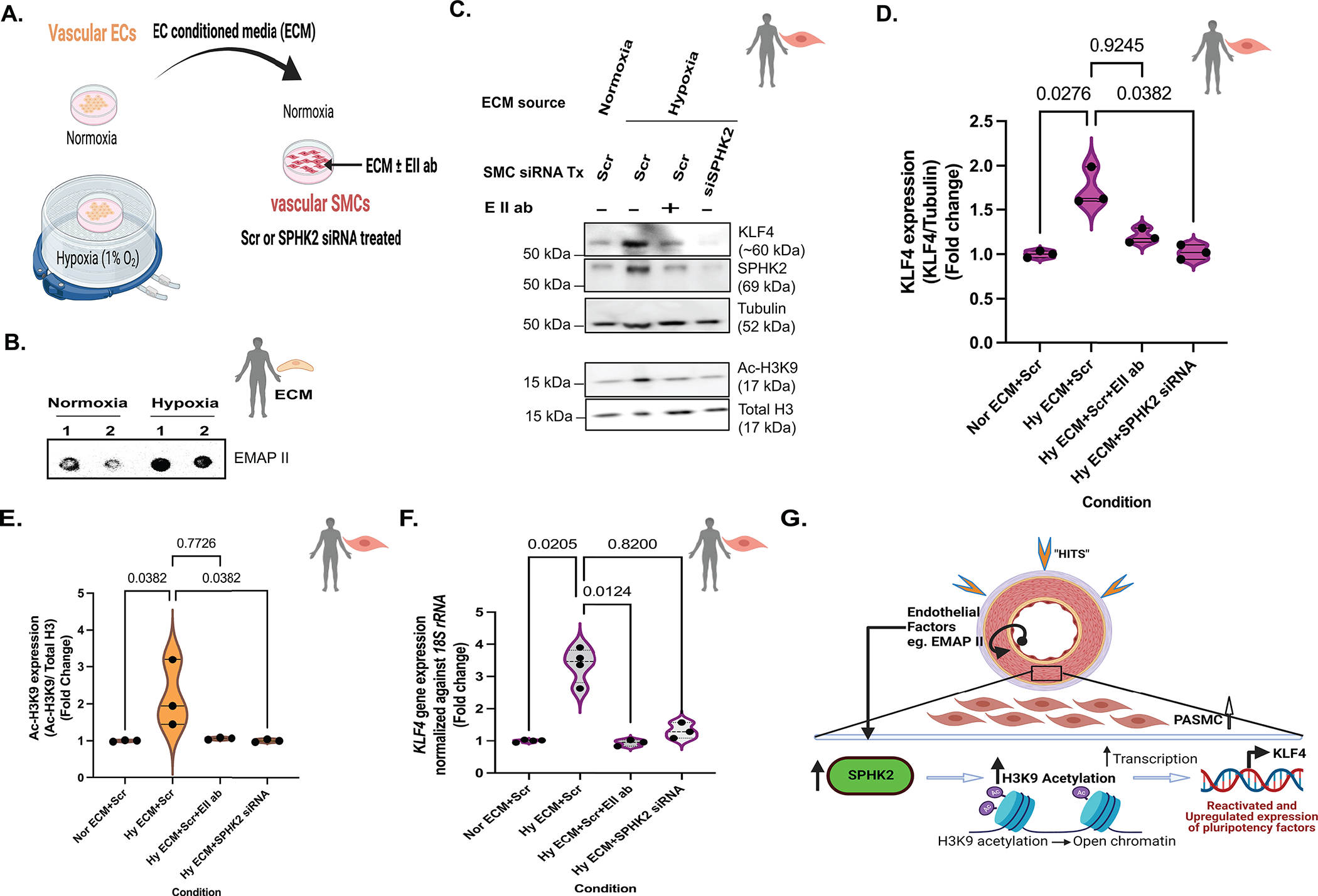
Results: We identified that Human PASMCs are vulnerable to the transcription-promoting epigenetic mediator histone acetylation resulting in alterations in transcription machinery and confirmed its pathological existence in PH:PASMC cells. We report that SPHK2 is elevated as much as 20-fold in iPAH lung tissue and is elevated in iPAH:PASMC cells. During PH pathogenesis, nuclear SPHK2 activates nuclear bioactive lipid S1P (sphingosine 1-phosphate) catalyzing enzyme and mediates transcription regulating histone H3K9 acetylation (acetyl histone H3 lysine 9 [Ac-H3K9]) through EMAP (endothelial monocyte activating polypeptide) II. In iPAH lungs, we identified a 4-fold elevation of the reversible epigenetic transcription modulator Ac-H3K9:H3 ratio. Loss of SPHK2 inhibited hypoxic-induced PH and Ac-H3K9 in mice. We discovered that pulmonary vascular endothelial cells are a priming factor of the EMAP II/SPHK2/S1P axis that alters the acetylome with a specificity for PASMC, through hyperacetylation of histone H3K9. Using cleavage under targets and release using nuclease, we further show that EMAP II-mediated SPHK2 has the potential to modify the local transcription machinery of pluripotency factor KLF4 (Krüppel-like factor 4) by hyperacetylating KLF4 Cis-regulatory elements while deletion and targeted inhibition of SPHK2 rescues transcription altering Ac-H3K9.
Conclusions: SPHK2 expression and its activation of the reversible histone H3K9 acetylation in human pulmonary artery smooth muscle cell represent new therapeutic targets that could mitigate PH vascular remodeling.
Keywords: chromatin; endothelial cells; epigenomics; hypertension, pulmonary.
Conflict of interest statement
Figures
References
-
- Wang L, Moonen JR, Cao A, Isobe S, Li CG, Tojais NF, Taylor S, Marciano DP, Chen PI, Gu M, Li D, Harper RL, El-Bizri N, Kim YM, Stankunas K, Rabinovitch M. Dysregulated Smooth Muscle Cell BMPR2–ARRB2 Axis Causes Pulmonary Hypertension. Circ Res 2023;132:545–564. doi:10.1161/CIRCRESAHA.121.320541. - DOI - PMC - PubMed
-
- Bisserier M, Mathiyalagan P, Zhang S, Elmastour F, Dorfmüller P, Humbert M, David G, Tarzami S, Weber T, Perros F, Sassi Y, Sahoo S, Hadri L. Regulation of the Methylation and Expression Levels of the BMPR2 Gene by SIN3a as a Novel Therapeutic Mechanism in Pulmonary Arterial Hypertension. Circulation 2021;144:52–73. doi:10.1161/CIRCULATIONAHA.120.047978. - DOI - PMC - PubMed
-
- Benincasa G, Maron BA, Affinito O, D’Alto M, Franzese M, Argiento P, Schiano C, Romeo E, Bontempo P, Golino P, Berrino L, Loscalzo J, Napoli C. Association Between Circulating CD4+ T Cell Methylation Signatures of Network-Oriented SOCS3 Gene and Hemodynamics in Patients Suffering Pulmonary Arterial Hypertension. J Cardiovasc Transl Res 2023;16:17–30. doi:10.1007/S12265-022-10294-1/FIGURES/6. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
