

Extending the reach of homology by using successive computational filters to find yeast pheromone genes
- PMID: 37699395
- PMCID: PMC10592104
- DOI: 10.1016/j.cub.2023.08.039
Extending the reach of homology by using successive computational filters to find yeast pheromone genes
Abstract
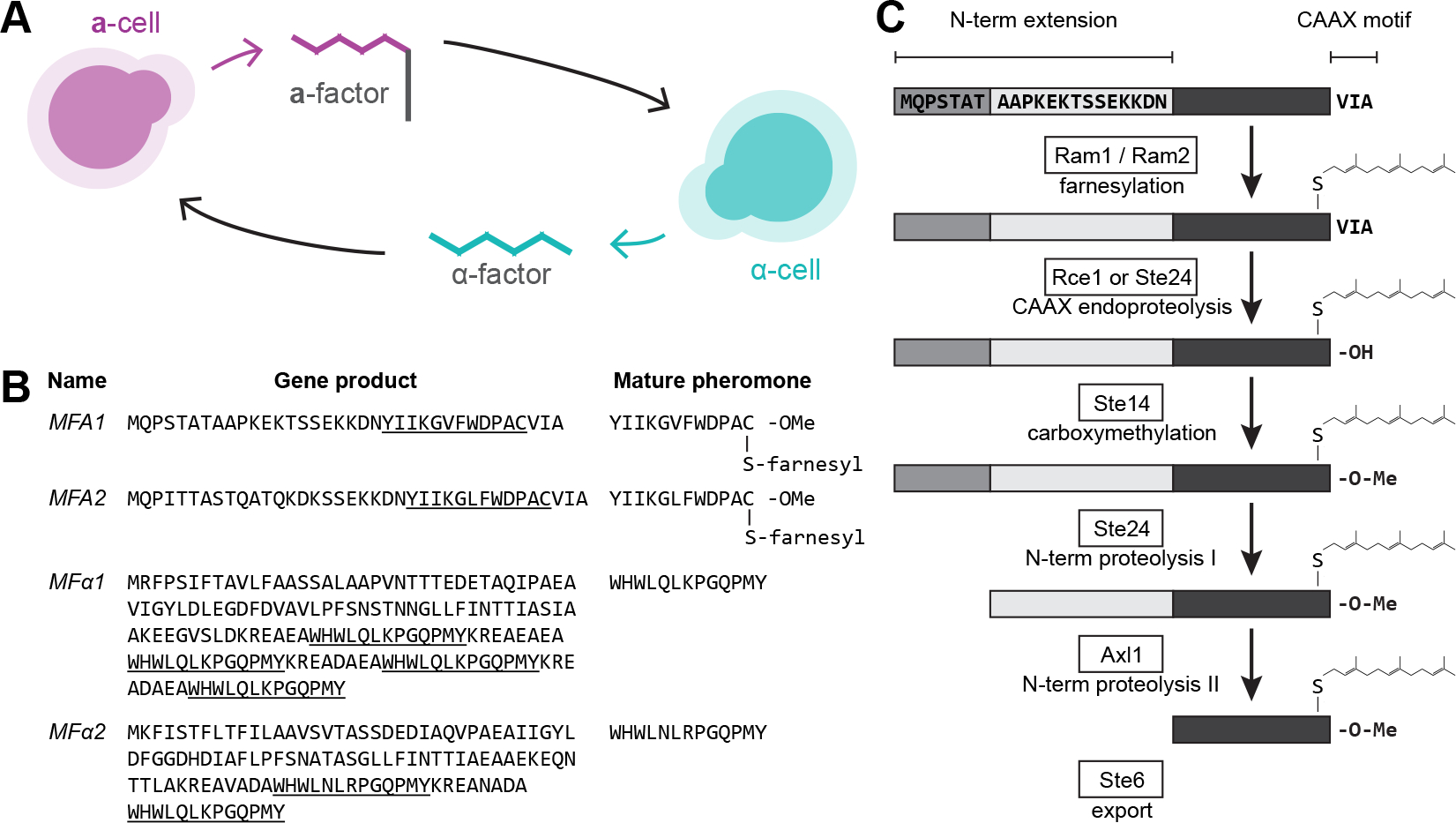
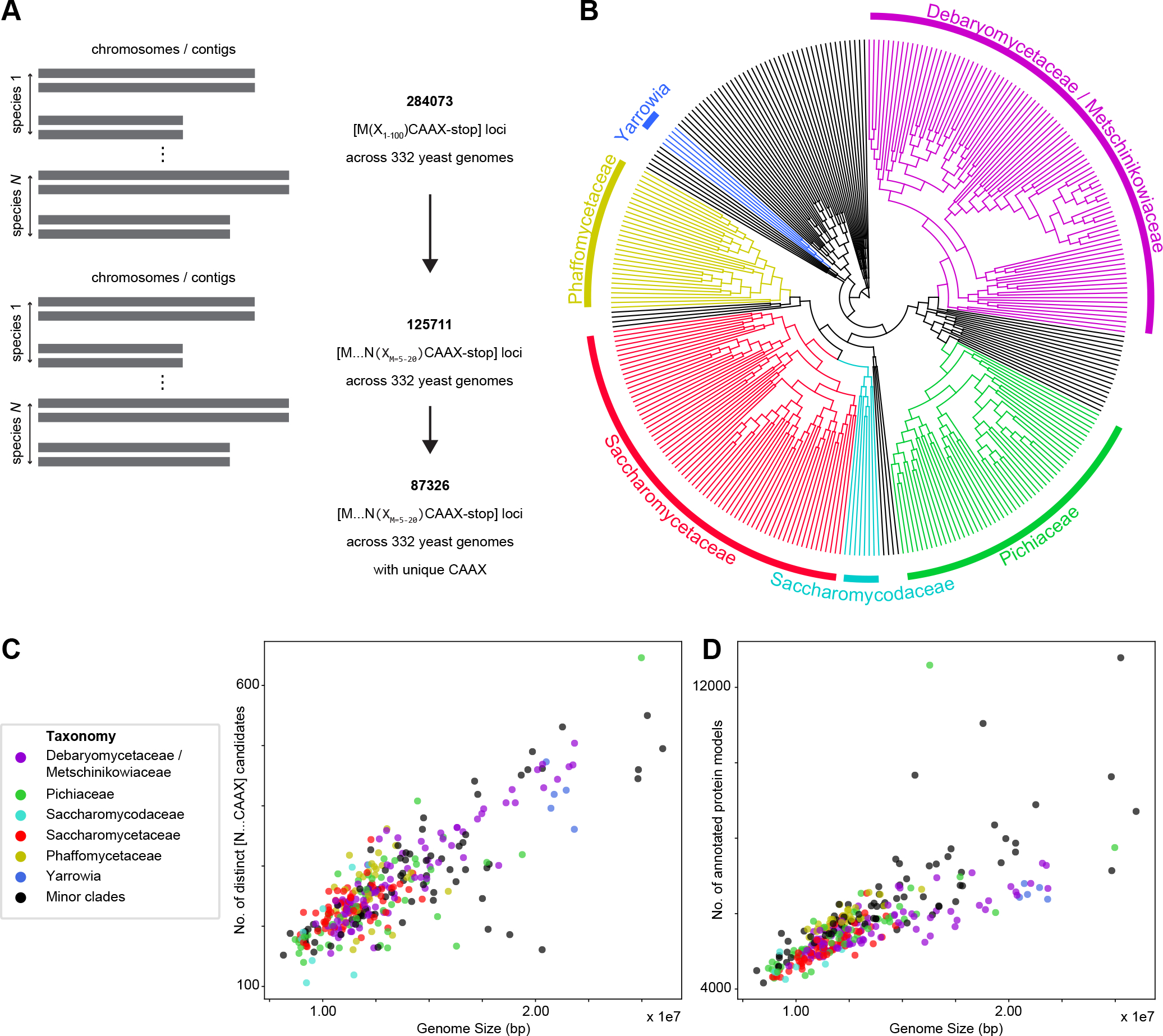
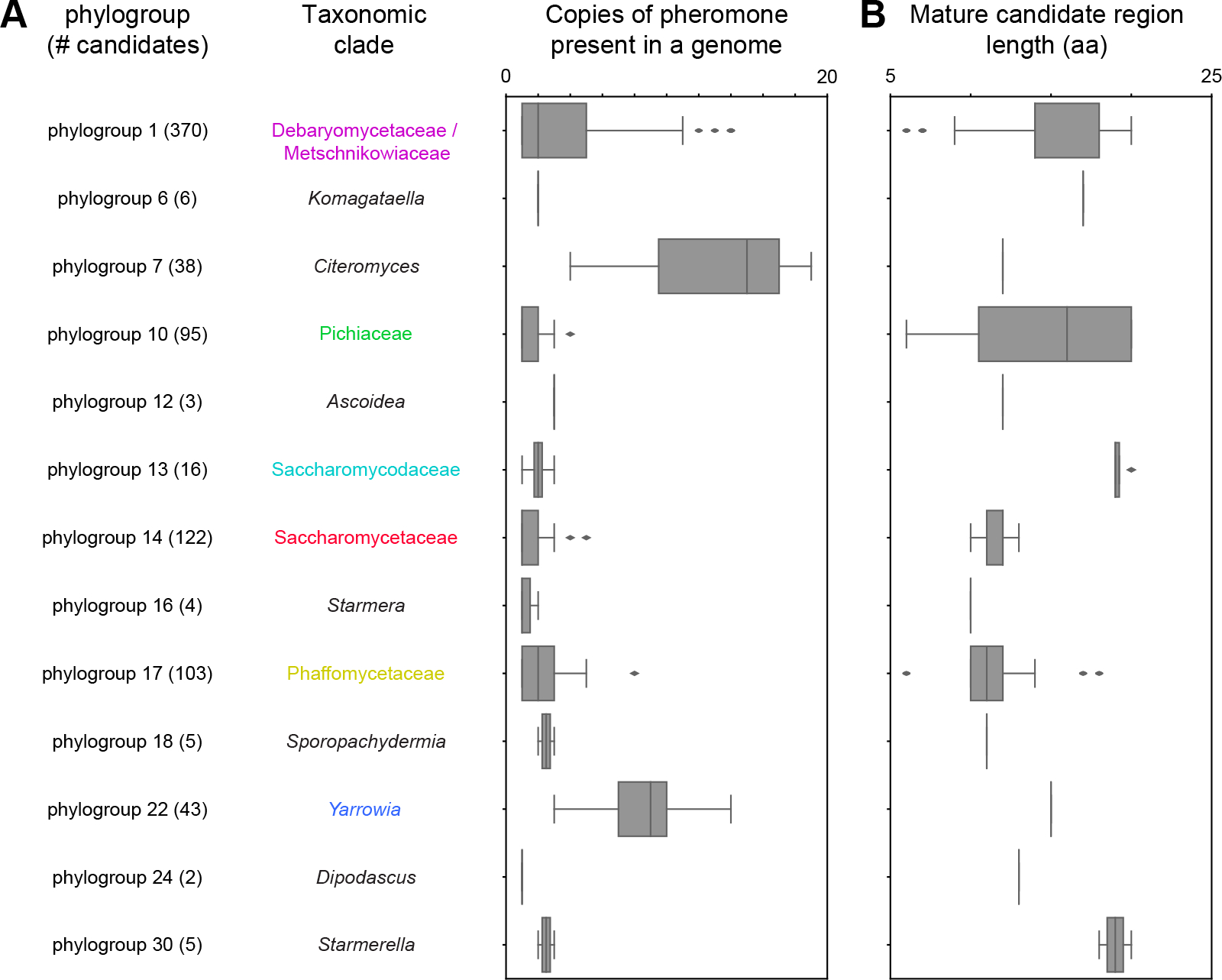
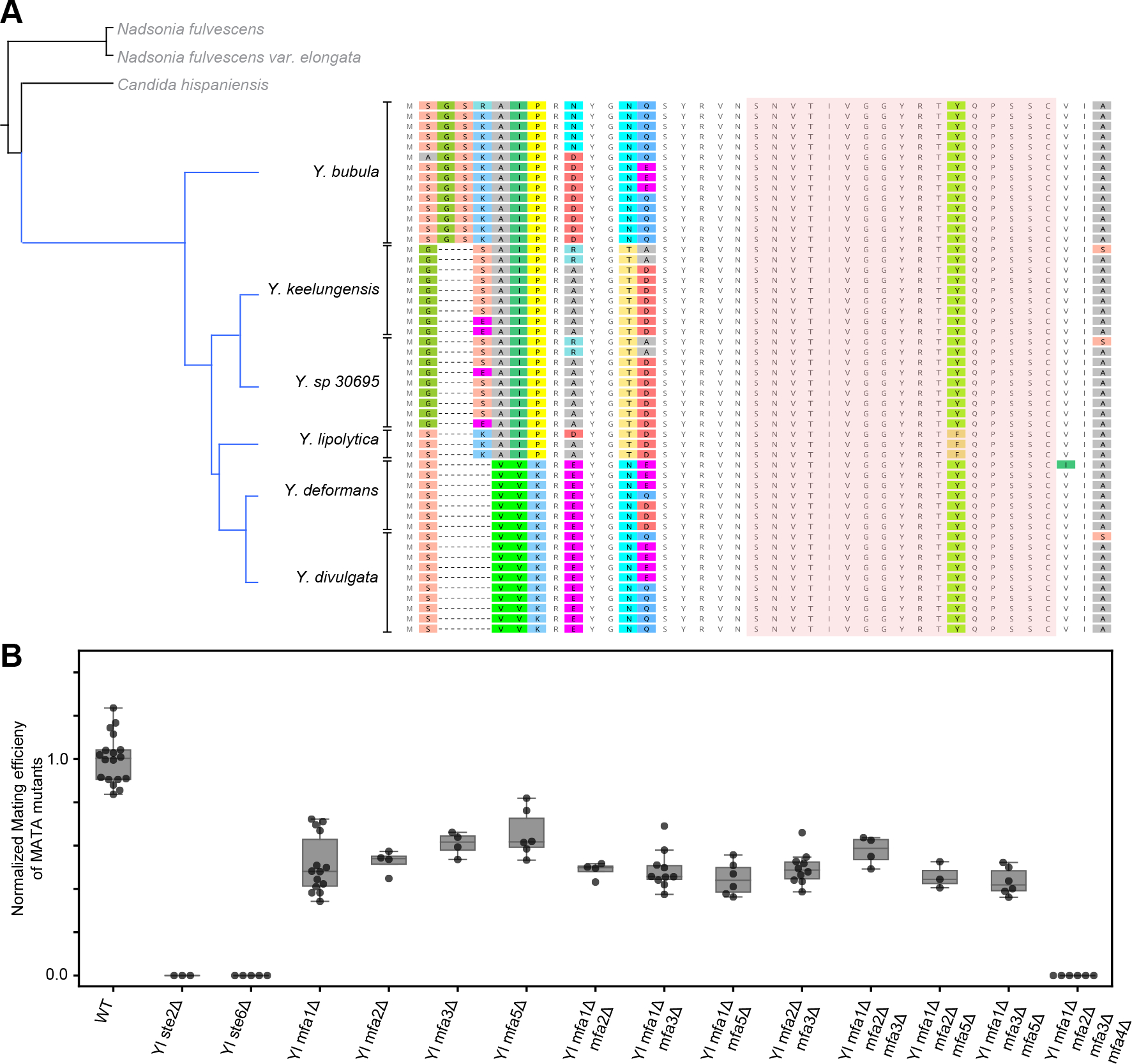
The mating of fungi depends on pheromones that mediate communication between two mating types. Most species use short peptides as pheromones, which are either unmodified (e.g., α-factor in Saccharomyces cerevisiae) or C-terminally farnesylated (e.g., a-factor in S. cerevisiae). Peptide pheromones have been found by genetics or biochemistry in a small number of fungi, but their short sequences and modest conservation make it impossible to detect homologous sequences in most species. To overcome this problem, we used a four-step computational pipeline to identify candidate a-factor genes in sequenced genomes of the Saccharomycotina, the fungal clade that contains most of the yeasts: we require that candidate genes have a C-terminal prenylation motif, are shorter than 100 amino acids long, and contain a proteolytic-processing motif upstream of the potential mature pheromone sequence and that closely related species contain highly conserved homologs of the potential mature pheromone sequence. Additional manual curation exploits the observation that many species carry more than one a-factor gene, encoding identical or nearly identical pheromones. From 332 Saccharomycotina genomes, we identified strong candidate pheromone genes in 241 genomes, covering 13 clades that are each separated from each other by at least 100 million years, the time required for evolution to remove detectable sequence homology among small pheromone genes. For one small clade, the Yarrowia, we demonstrated that our algorithm found the a-factor genes: deleting all four related genes in the a-mating type of Yarrowia lipolytica prevents mating.
Keywords: Yarrowia; gene annotation; pheromones; small peptides; yeast mating.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

Similar articles
-
Point mutations identify a conserved region of the saccharomyces cerevisiae AFR1 gene that is essential for both the pheromone signaling and morphogenesis functions.Genetics. 2000 May;155(1):43-55. doi: 10.1093/genetics/155.1.43. Genetics. 2000. PMID: 10790383 Free PMC article.
-
Asymmetry in sexual pheromones is not required for ascomycete mating.Curr Biol. 2011 Aug 23;21(16):1337-46. doi: 10.1016/j.cub.2011.06.054. Epub 2011 Aug 11. Curr Biol. 2011. PMID: 21835624 Free PMC article.
-
Komagataella phaffii YPS1-5 encodes the alpha-factor degrading protease Bar1.FEMS Yeast Res. 2020 May 1;20(3):foaa024. doi: 10.1093/femsyr/foaa024. FEMS Yeast Res. 2020. PMID: 32374383
-
Pheromone Response and Mating Behavior in Fission Yeast.Microbiol Mol Biol Rev. 2022 Dec 21;86(4):e0013022. doi: 10.1128/mmbr.00130-22. Epub 2022 Dec 5. Microbiol Mol Biol Rev. 2022. PMID: 36468849 Free PMC article. Review.
-
Basidiomycete mating type genes and pheromone signaling.Eukaryot Cell. 2010 Jun;9(6):847-59. doi: 10.1128/EC.00319-09. Epub 2010 Feb 26. Eukaryot Cell. 2010. PMID: 20190072 Free PMC article. Review.
Cited by
-
Needles in fungal haystacks: Discovery of a putative a-factor pheromone and a unique mating strategy in the Leotiomycetes.PLoS One. 2023 Oct 12;18(10):e0292619. doi: 10.1371/journal.pone.0292619. eCollection 2023. PLoS One. 2023. PMID: 37824487 Free PMC article.
-
Structure and number of mating pheromone genes is closely linked to sexual reproductive strategy in Huntiella.BMC Genomics. 2023 May 13;24(1):261. doi: 10.1186/s12864-023-09355-9. BMC Genomics. 2023. PMID: 37179314 Free PMC article.
References
-
- Lücking R, Huhndorf S, Pfister DH, Plata ER, and Lumbsch HT (2017). Fungi evolved right on track. Mycologia 101, 810–822. - PubMed
-
- Kurjan J, and Herskowitz I (1982). Structure of a yeast pheromone gene (MF alpha): a putative alpha-factor precursor contains four tandem copies of mature alpha-factor. Cell 30, 933–943. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
