

Inferring the Effects of Protein Variants on Protein-Protein Interactions with Interpretable Transformer Representations
- PMID: 37701056
- PMCID: PMC10494974
- DOI: 10.34133/research.0219
Inferring the Effects of Protein Variants on Protein-Protein Interactions with Interpretable Transformer Representations
Abstract
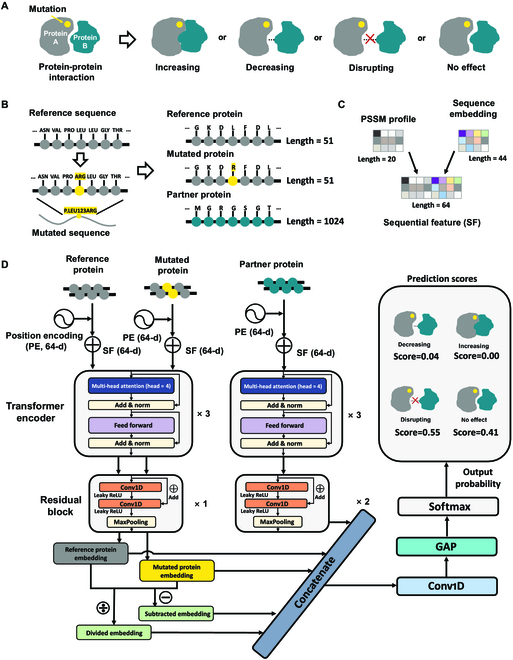
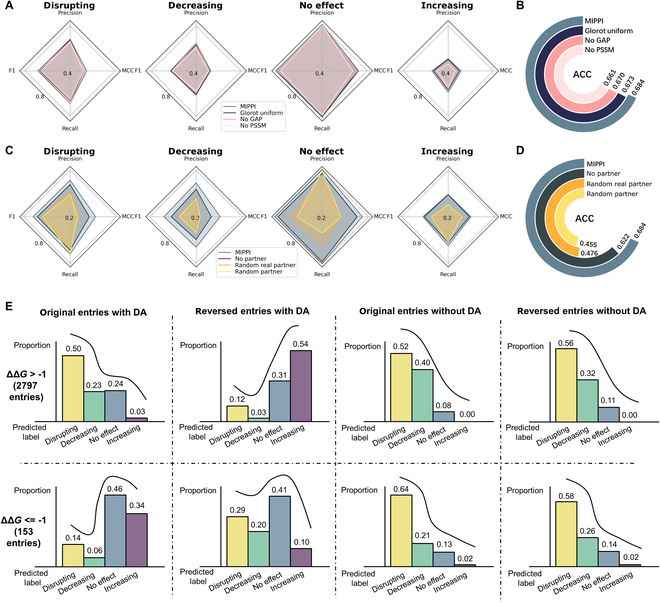
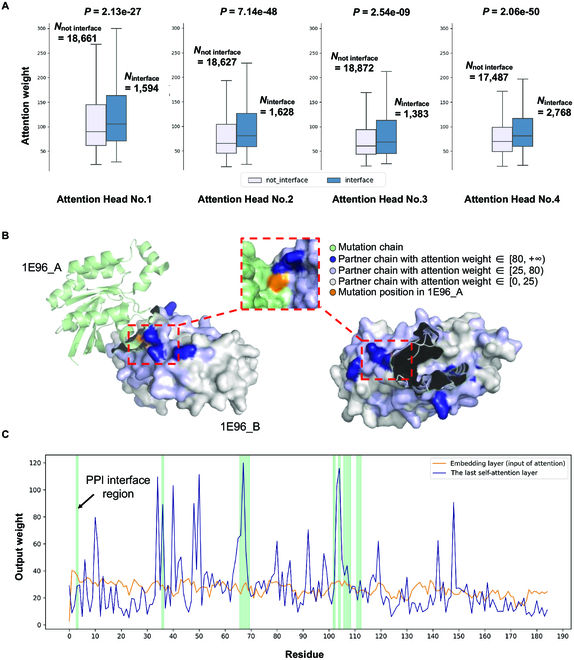
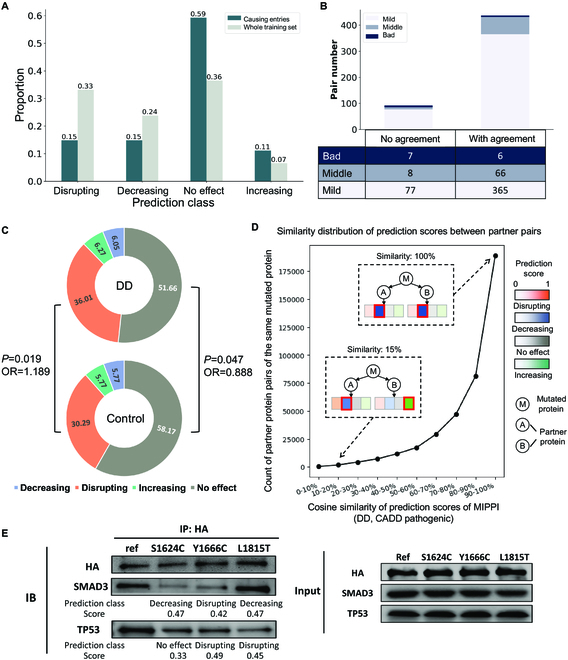
Identifying pathogenetic variants and inferring their impact on protein-protein interactions sheds light on their functional consequences on diseases. Limited by the availability of experimental data on the consequences of protein interaction, most existing methods focus on building models to predict changes in protein binding affinity. Here, we introduced MIPPI, an end-to-end, interpretable transformer-based deep learning model that learns features directly from sequences by leveraging the interaction data from IMEx. MIPPI was specifically trained to determine the types of variant impact (increasing, decreasing, disrupting, and no effect) on protein-protein interactions. We demonstrate the accuracy of MIPPI and provide interpretation through the analysis of learned attention weights, which exhibit correlations with the amino acids interacting with the variant. Moreover, we showed the practicality of MIPPI in prioritizing de novo mutations associated with complex neurodevelopmental disorders and the potential to determine the pathogenic and driving mutations. Finally, we experimentally validated the functional impact of several variants identified in patients with such disorders. Overall, MIPPI emerges as a versatile, robust, and interpretable model, capable of effectively predicting mutation impacts on protein-protein interactions and facilitating the discovery of clinically actionable variants.
Copyright © 2023 Zhe Liu et al.
Figures
Similar articles
-
Inferring the molecular and phenotypic impact of amino acid variants with MutPred2.Nat Commun. 2020 Nov 20;11(1):5918. doi: 10.1038/s41467-020-19669-x. Nat Commun. 2020. PMID: 33219223 Free PMC article.
-
ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction.Nat Methods. 2022 Jun;19(6):730-739. doi: 10.1038/s41592-022-01490-7. Epub 2022 May 30. Nat Methods. 2022. PMID: 35637310
-
DTITR: End-to-end drug-target binding affinity prediction with transformers.Comput Biol Med. 2022 Aug;147:105772. doi: 10.1016/j.compbiomed.2022.105772. Epub 2022 Jun 21. Comput Biol Med. 2022. PMID: 35777085
-
A deep learning model for characterizing protein-RNA interactions from sequences at single-base resolution.Patterns (N Y). 2025 Jan 10;6(1):101150. doi: 10.1016/j.patter.2024.101150. eCollection 2025 Jan 10. Patterns (N Y). 2025. PMID: 39896261 Free PMC article.
-
Do it the transformer way: A comprehensive review of brain and vision transformers for autism spectrum disorder diagnosis and classification.Comput Biol Med. 2023 Dec;167:107667. doi: 10.1016/j.compbiomed.2023.107667. Epub 2023 Nov 3. Comput Biol Med. 2023. PMID: 37939407 Review.
Cited by
-
Molecular Merged Hypergraph Neural Network for Explainable Solvation Gibbs Free Energy Prediction.Research (Wash D C). 2025 Aug 15;8:0740. doi: 10.34133/research.0740. eCollection 2025. Research (Wash D C). 2025. PMID: 40822120 Free PMC article.
-
ProstaNet: A Novel Geometric Vector Perceptrons-Graph Neural Network Algorithm for Protein Stability Prediction in Single- and Multiple-Point Mutations with Experimental Validation.Research (Wash D C). 2025 Apr 15;8:0674. doi: 10.34133/research.0674. eCollection 2025. Research (Wash D C). 2025. PMID: 40235597 Free PMC article.
-
S2ALM: Sequence-Structure Pre-trained Large Language Model for Comprehensive Antibody Representation Learning.Research (Wash D C). 2025 Aug 19;8:0721. doi: 10.34133/research.0721. eCollection 2025. Research (Wash D C). 2025. PMID: 40837877 Free PMC article.
References
-
- Matos B, Howl J, Jerónimo C, Fardilha M. The disruption of protein-protein interactions as a therapeutic strategy for prostate cancer. Pharmacol Res. 2020;161:105145. - PubMed
-
- Cummings CG, Hamilton AD. Disrupting protein–protein interactions with non-peptidic, small molecule α-helix mimetics. Curr Opin Chem Biol. 2010;14(3):341–346. - PubMed
LinkOut - more resources
Full Text Sources
