

Identification and validation of key biomarkers based on RNA methylation genes in sepsis
- PMID: 37701433
- PMCID: PMC10493392
- DOI: 10.3389/fimmu.2023.1231898
Identification and validation of key biomarkers based on RNA methylation genes in sepsis
Abstract
Background: RNA methylation is closely involved in immune regulation, but its role in sepsis remains unknown. Here, we aim to investigate the role of RNA methylation-associated genes (RMGs) in classifying and diagnosing of sepsis.
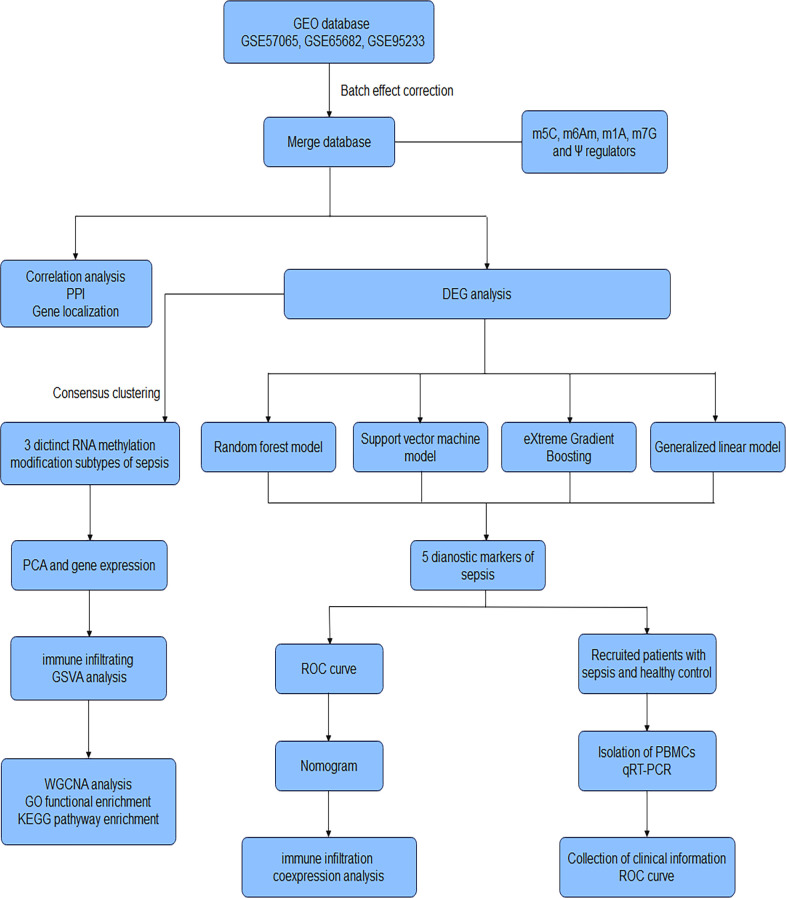
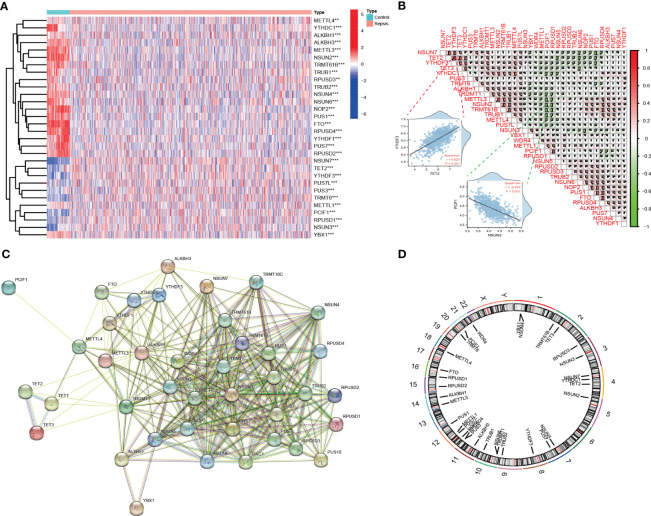
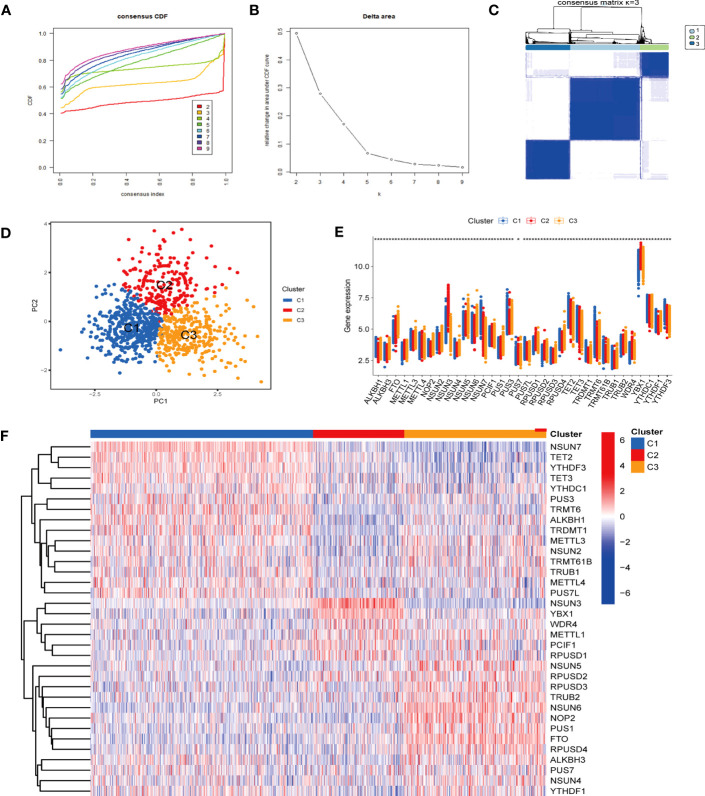
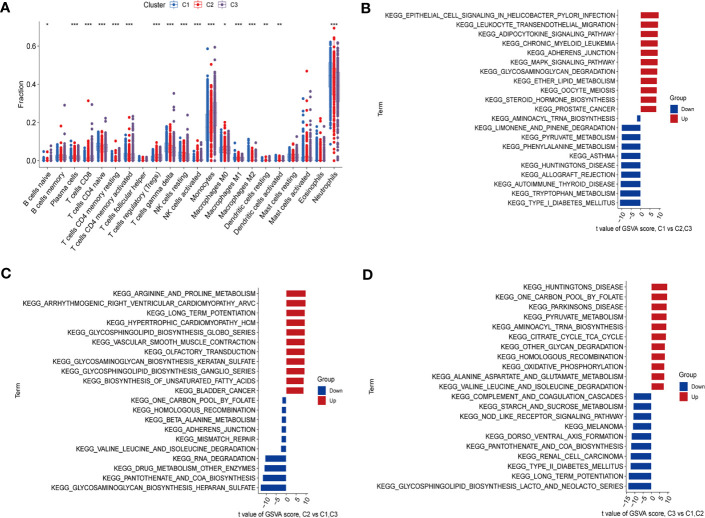
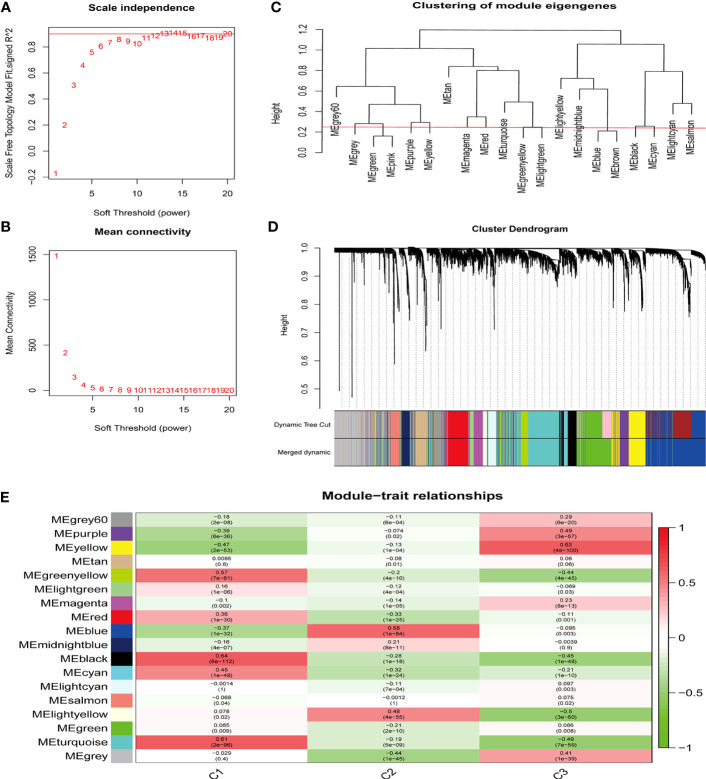
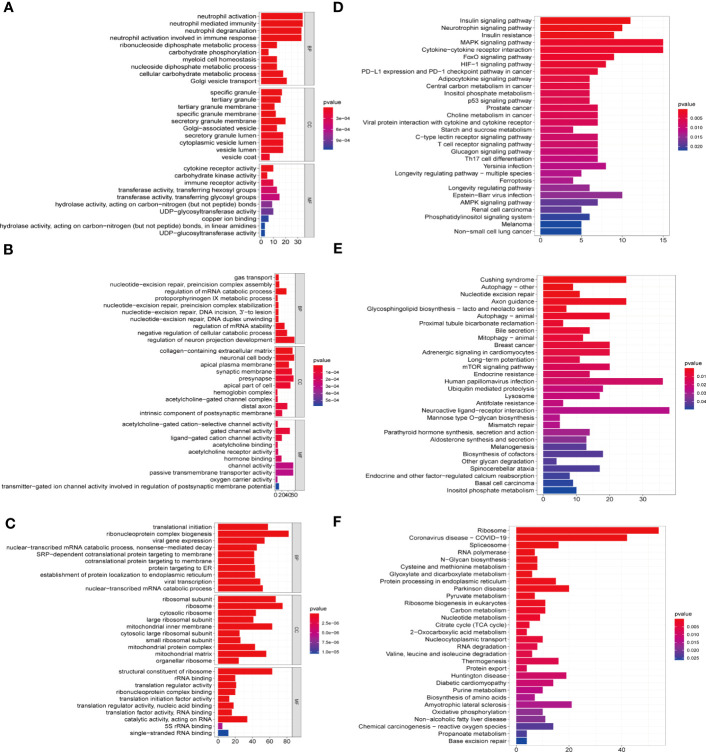
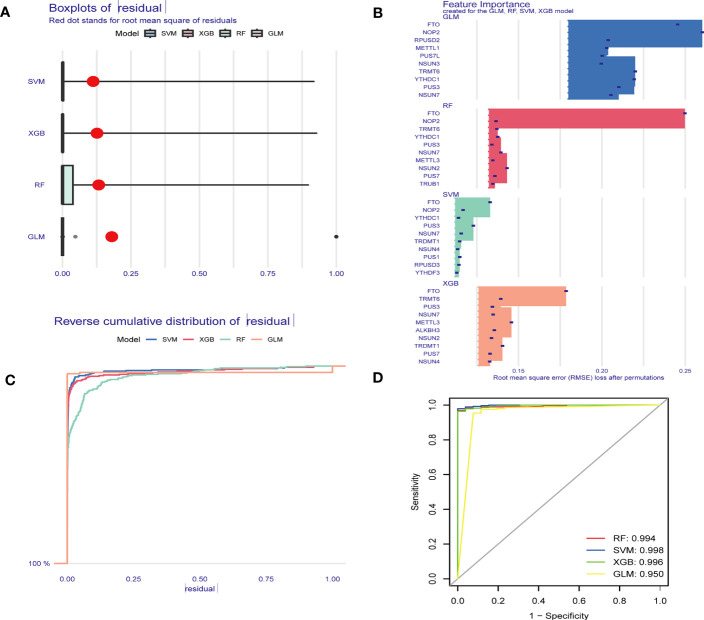
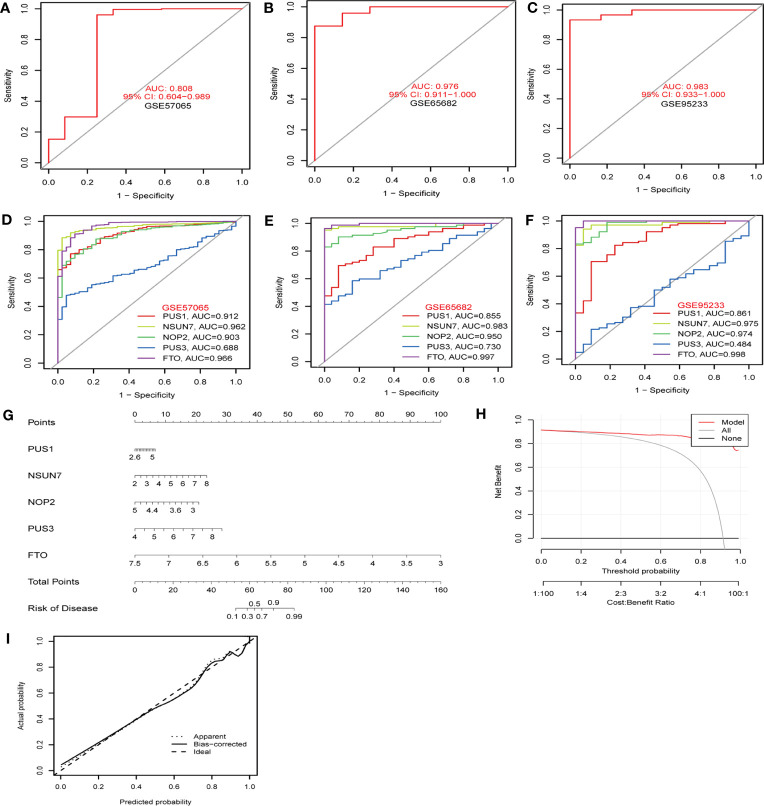
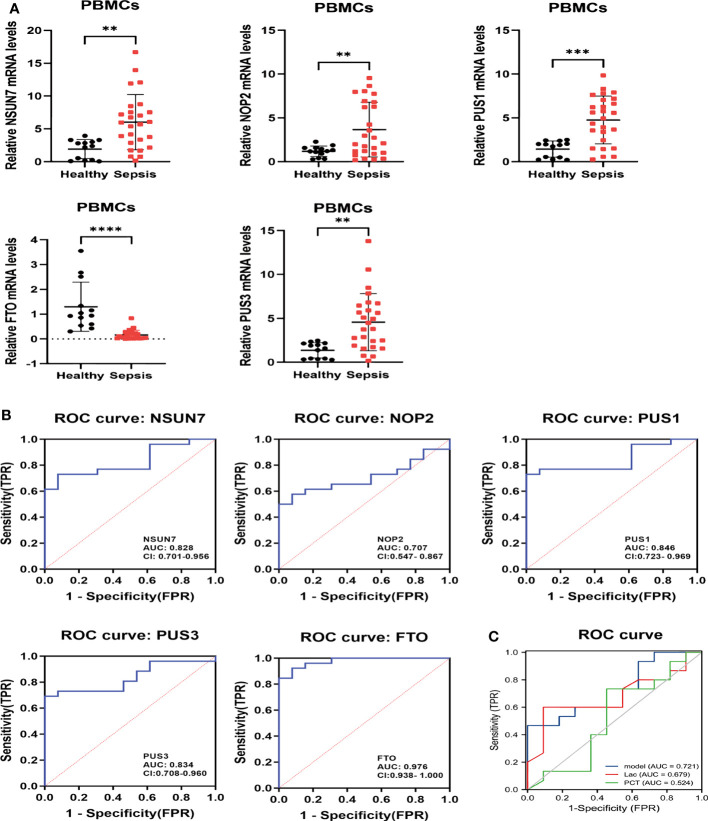
Methods: Five types of RMGs (m1A, m5C, m6Am, m7G and Ψ) were used to identify sepsis subgroups based on gene expression profile data obtained from the GEO database (GSE57065, GSE65682, and GSE95233). Unsupervised clustering analysis was used to identify distinct RNA modification subtypes. The CIBERSORT, WGCNA, GO and KEGG analysis were performed to explore immune infiltration pattern and biological function of each cluster. RF, SVM, XGB, and GLM algorithm were applied to identify the diagnostic RMGs in sepsis. Finally, the expression levels of the five key RMGs were verified by collecting PBMCs from septic patients using qRT-PCR, and their diagnostic efficacy for sepsis was verified in combination with clinical data using ROC analysis.
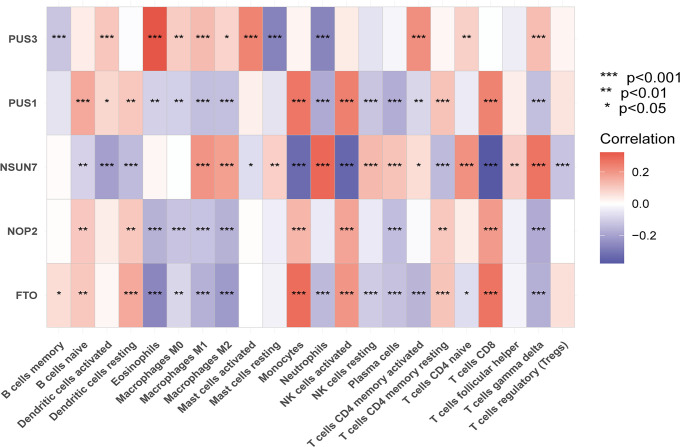
Results: Sepsis was divided into three subtypes (cluster 1 to 3). Cluster 1 highly expressed NSUN7 and TRMT6, with the characteristic of neutrophil activation and upregulation of MAPK signaling pathways. Cluster 2 highly expressed NSUN3, and was featured by the regulation of mRNA stability and amino acid metabolism. NSUN5 and NSUN6 were upregulated in cluster 3 which was involved in ribonucleoprotein complex biogenesis and carbohydrate metabolism pathways. In addition, we identified that five RMGs (NSUN7, NOP2, PUS1, PUS3 and FTO) could function as biomarkers for clinic diagnose of sepsis. For validation, we determined that the relative expressions of NSUN7, NOP2, PUS1 and PUS3 were upregulated, while FTO was downregulated in septic patients. The area under the ROC curve (AUC) of NSUN7, NOP2, PUS1, PUS3 and FTO was 0.828, 0.707, 0.846, 0.834 and 0.976, respectively.
Conclusions: Our study uncovered that dysregulation of RNA methylation genes (m1A, m5C, m6Am, m7G and Ψ) was closely involved in the pathogenesis of sepsis, providing new insights into the classification of sepsis endotypes. We also revealed that five hub RMGs could function as novel diagnostic biomarkers and potential targets for treatment.
Keywords: RNA methylation; biomarkers; machine learning; sepsis; unsupervised clustering.
Copyright © 2023 Zhang, Bao, Cui, Wang, Ji, Jing, Zhou, Chen and Tang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ferrer R, Martin-Loeches I, Phillips G, Osborn TM, Townsend S, Dellinger RP, et al. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: results from a guideline-based performance improvement program. Crit Care Med (2014) 42:1749–55. doi: 10.1097/ccm.0000000000000330 - DOI - PubMed
-
- de Grooth HJ, Postema J, Loer SA, Parienti JJ, Oudemans-van Straaten HM, Girbes AR. Unexplained mortality differences between septic shock trials: a systematic analysis of population characteristics and control-group mortality rates. Intensive Care Med (2018) 44:311–22. doi: 10.1007/s00134-018-5134-8 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical