

When will RNA get its AlphaFold moment?
- PMID: 37702120
- PMCID: PMC10570031
- DOI: 10.1093/nar/gkad726
When will RNA get its AlphaFold moment?
Abstract
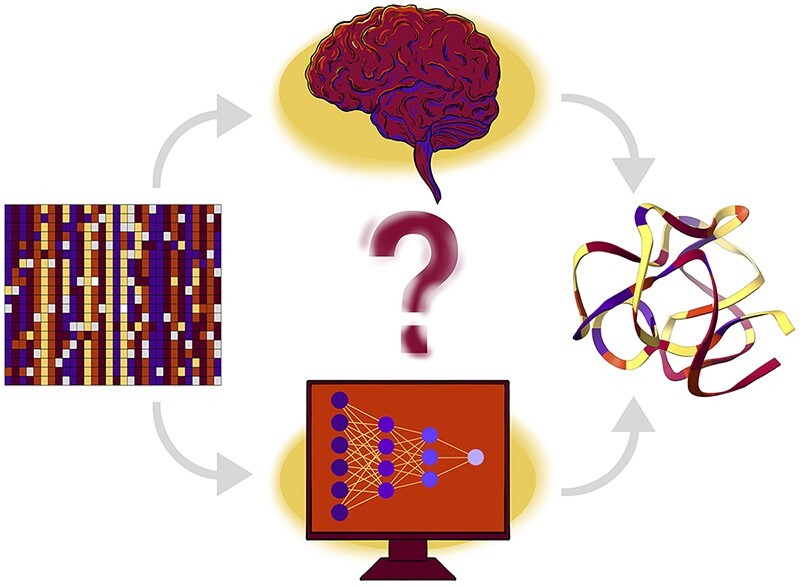
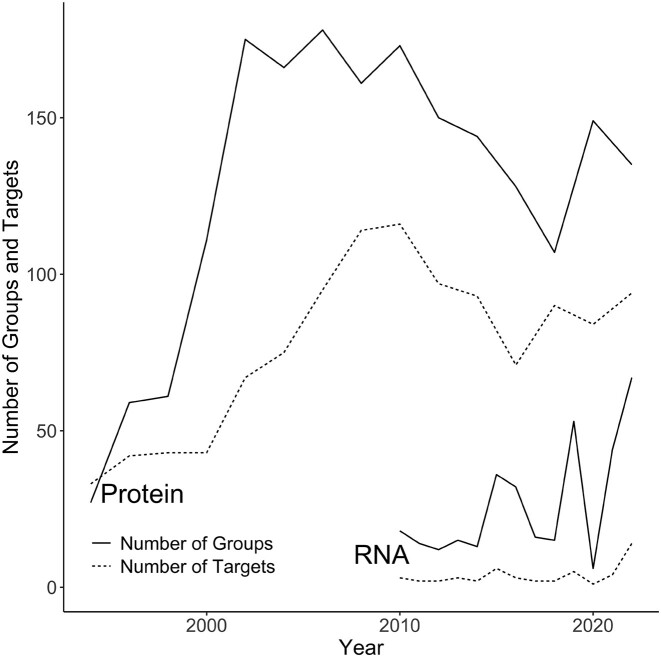
The protein structure prediction problem has been solved for many types of proteins by AlphaFold. Recently, there has been considerable excitement to build off the success of AlphaFold and predict the 3D structures of RNAs. RNA prediction methods use a variety of techniques, from physics-based to machine learning approaches. We believe that there are challenges preventing the successful development of deep learning-based methods like AlphaFold for RNA in the short term. Broadly speaking, the challenges are the limited number of structures and alignments making data-hungry deep learning methods unlikely to succeed. Additionally, there are several issues with the existing structure and sequence data, as they are often of insufficient quality, highly biased and missing key information. Here, we discuss these challenges in detail and suggest some steps to remedy the situation. We believe that it is possible to create an accurate RNA structure prediction method, but it will require solving several data quality and volume issues, usage of data beyond simple sequence alignments, or the development of new less data-hungry machine learning methods.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
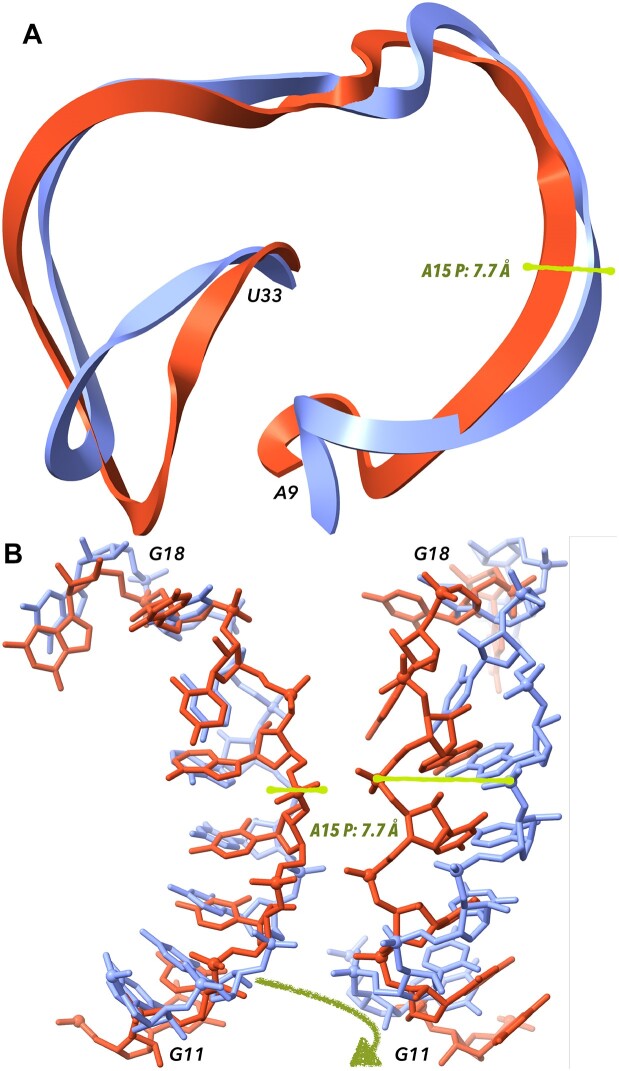
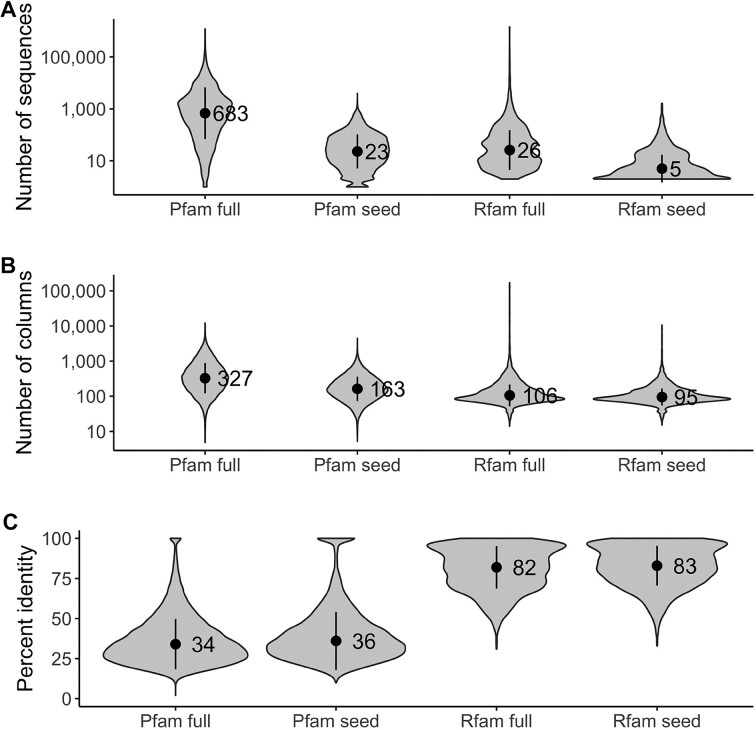
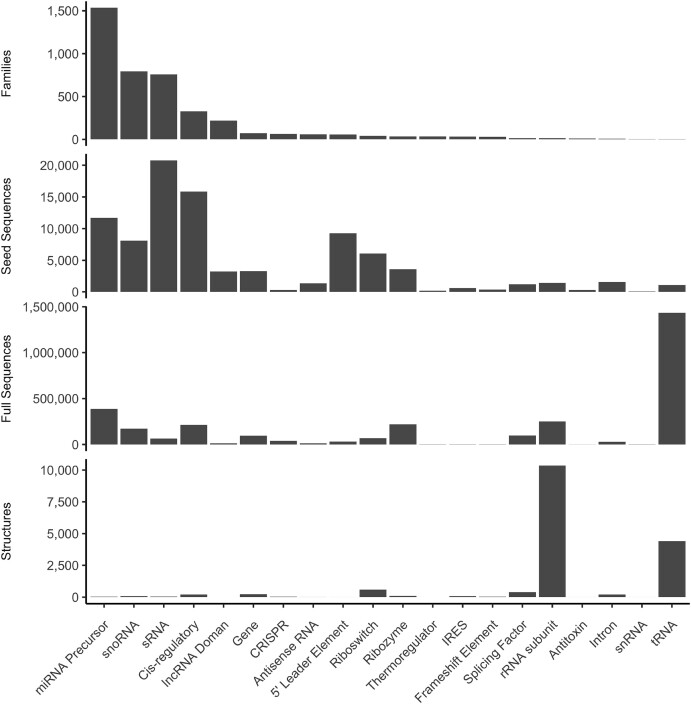
References
-
- Cech T.R., Steitz J.A., Atkins J.F.. RNA worlds: New tools for deep exploration. 2019; NY: Cold Spring Harbor Laboratory Press.
-
- Matzov D., Bashan A., Yonath A.. A bright future for antibiotics. Ann. Rev. Biochem. 2017; 86:567–583. - PubMed
-
- n.a. Big pharma craves slice of AI-based RNA drug discovery. Nat. Biotechnol. 2023; 41:305. - PubMed
-
- Tishchenko S., Kostareva O., Gabdulkhakov A., Mikhaylina A., Nikonova E., Nevskaya N., Sarskikh A., Piendl W., Garber M., Nikonov S.. Protein–RNA affinity of ribosomal protein L1 mutants does not correlate with the number of intermolecular interactions. Acta Crystallogr. D. 2015; 71:376–386. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
