

Clustering predicted structures at the scale of the known protein universe
- PMID: 37704730
- PMCID: PMC10584675
- DOI: 10.1038/s41586-023-06510-w
Clustering predicted structures at the scale of the known protein universe
Abstract
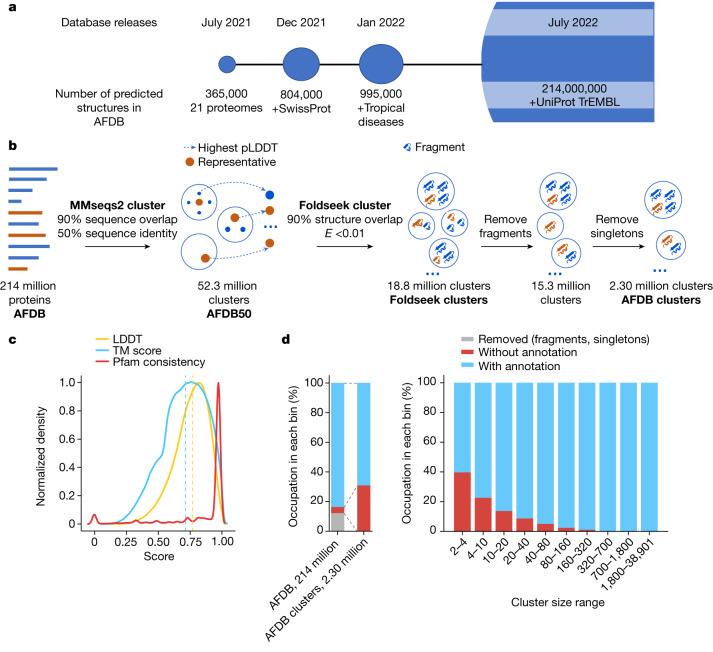
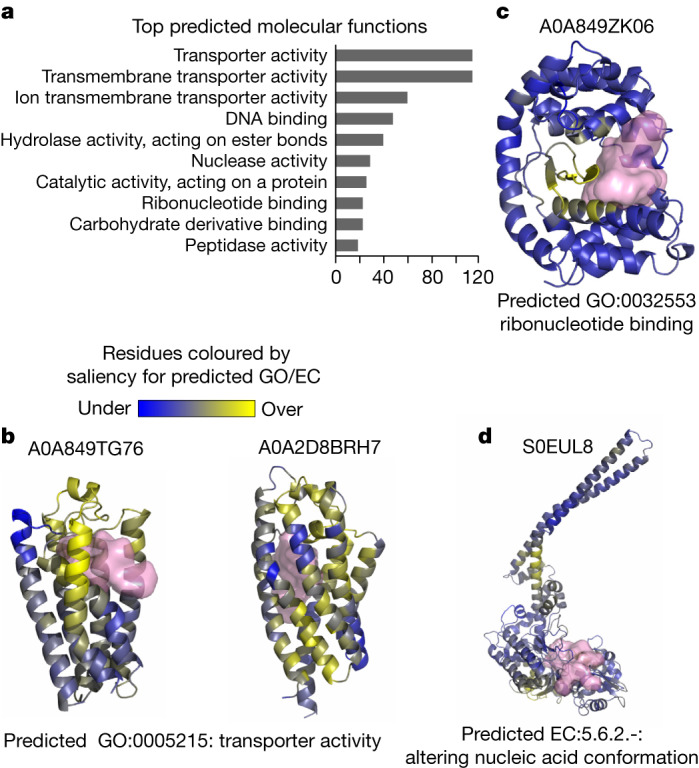
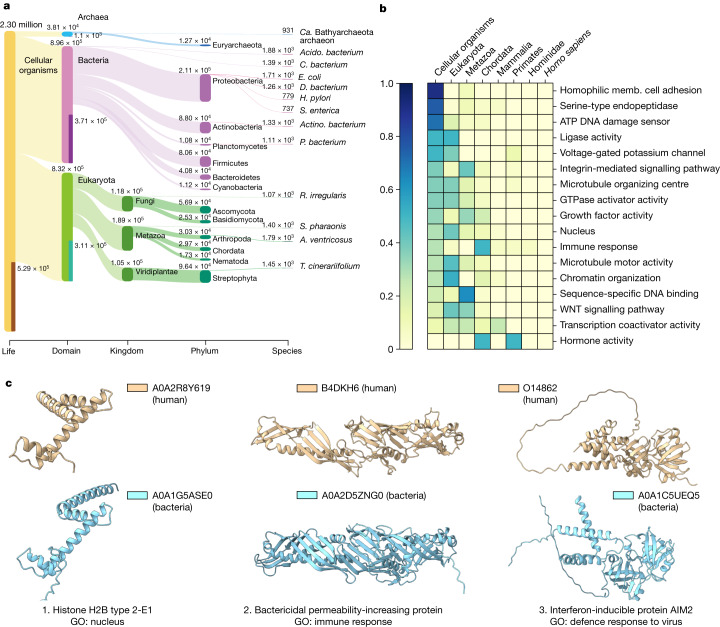
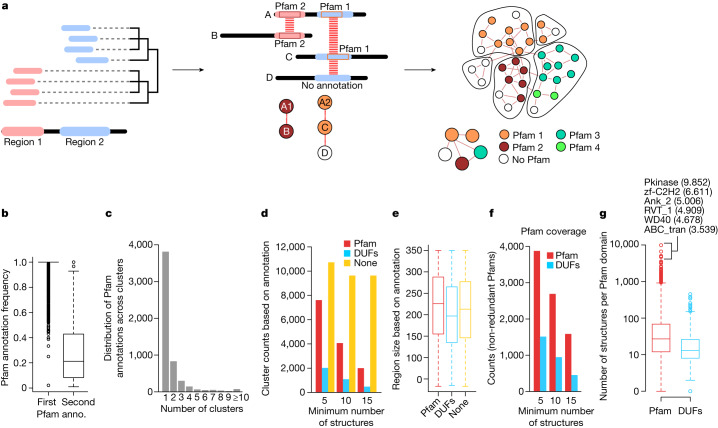
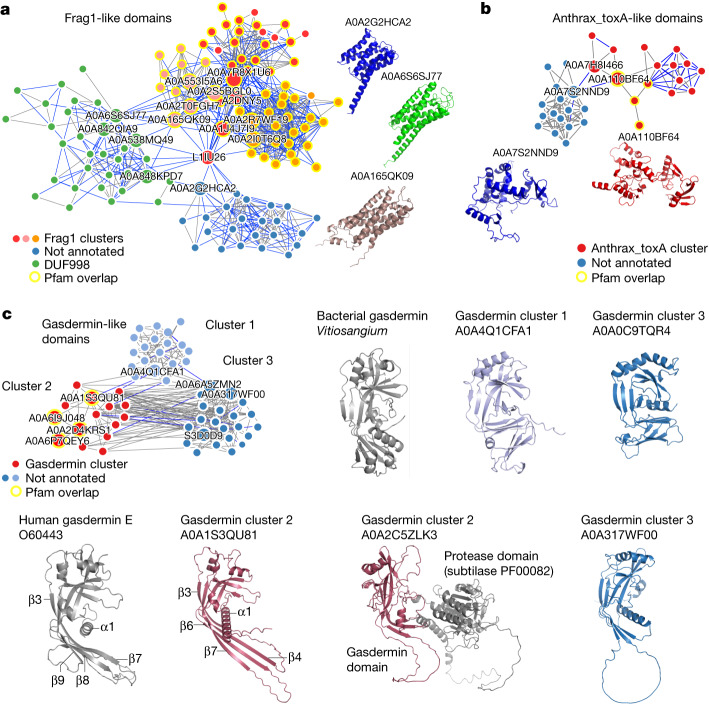
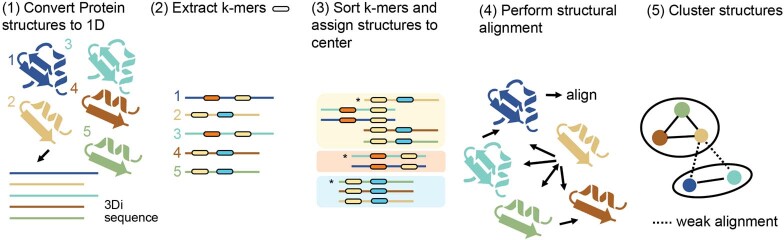
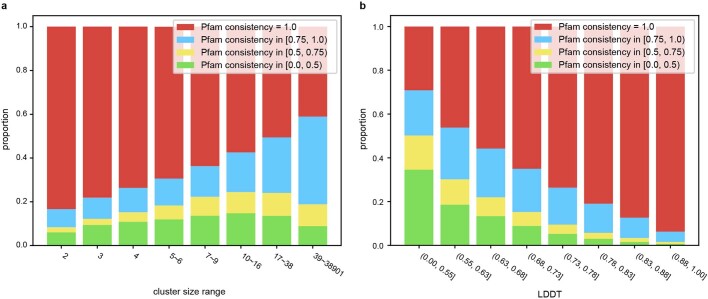
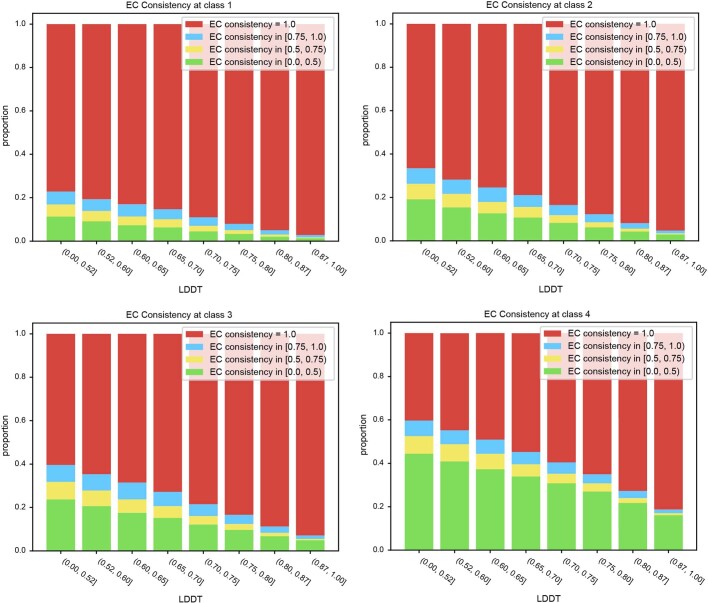
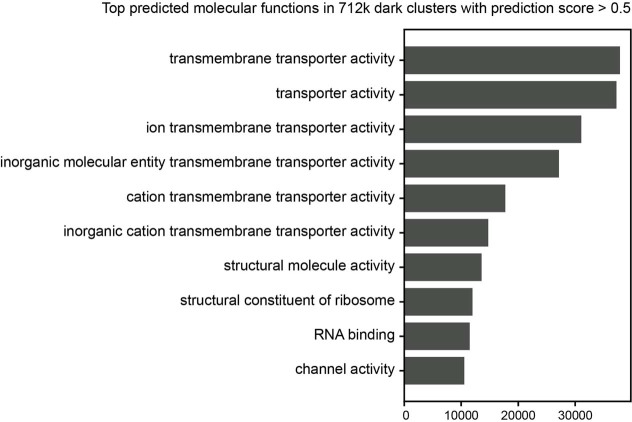
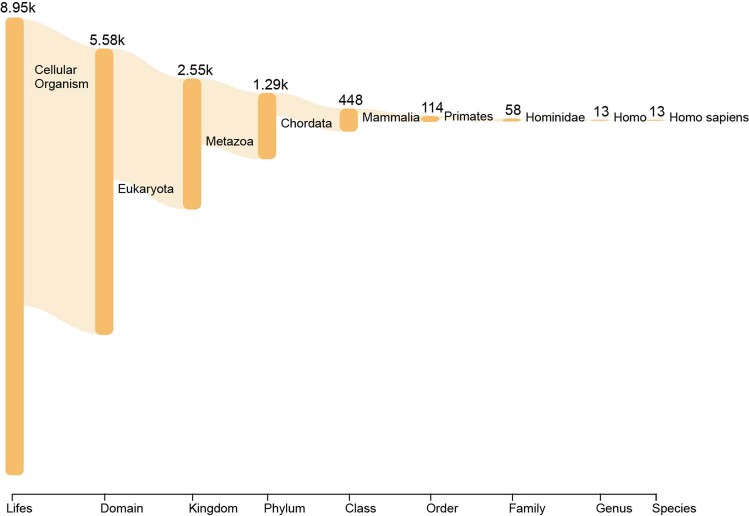
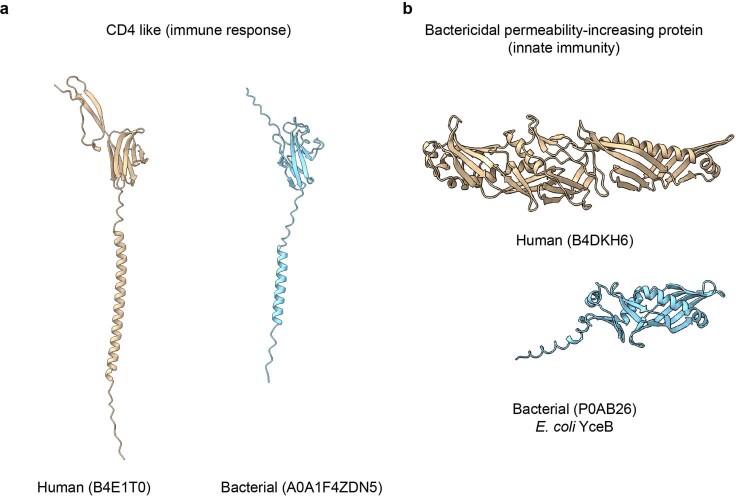
Proteins are key to all cellular processes and their structure is important in understanding their function and evolution. Sequence-based predictions of protein structures have increased in accuracy1, and over 214 million predicted structures are available in the AlphaFold database2. However, studying protein structures at this scale requires highly efficient methods. Here, we developed a structural-alignment-based clustering algorithm-Foldseek cluster-that can cluster hundreds of millions of structures. Using this method, we have clustered all of the structures in the AlphaFold database, identifying 2.30 million non-singleton structural clusters, of which 31% lack annotations representing probable previously undescribed structures. Clusters without annotation tend to have few representatives covering only 4% of all proteins in the AlphaFold database. Evolutionary analysis suggests that most clusters are ancient in origin but 4% seem to be species specific, representing lower-quality predictions or examples of de novo gene birth. We also show how structural comparisons can be used to predict domain families and their relationships, identifying examples of remote structural similarity. On the basis of these analyses, we identify several examples of human immune-related proteins with putative remote homology in prokaryotic species, illustrating the value of this resource for studying protein function and evolution across the tree of life.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Large-scale clustering of AlphaFold2 3D models shines light on the structure and function of proteins.Mol Cell. 2023 Nov 16;83(22):3950-3952. doi: 10.1016/j.molcel.2023.10.039. Epub 2023 Nov 16. Mol Cell. 2023. PMID: 37977115
References
-
- Terwilliger, T. C. et al. AlphaFold predictions: great hypotheses but no match for experiment. Preprint at bioRxiv10.1101/2022.11.21.517405 (2022).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
