

Treating Semiempirical Hamiltonians as Flexible Machine Learning Models Yields Accurate and Interpretable Results
- PMID: 37705220
- PMCID: PMC10536991
- DOI: 10.1021/acs.jctc.3c00491
Treating Semiempirical Hamiltonians as Flexible Machine Learning Models Yields Accurate and Interpretable Results
Abstract
Quantum chemistry provides chemists with invaluable information, but the high computational cost limits the size and type of systems that can be studied. Machine learning (ML) has emerged as a means to dramatically lower the cost while maintaining high accuracy. However, ML models often sacrifice interpretability by using components such as the artificial neural networks of deep learning that function as black boxes. These components impart the flexibility needed to learn from large volumes of data but make it difficult to gain insight into the physical or chemical basis for the predictions. Here, we demonstrate that semiempirical quantum chemical (SEQC) models can learn from large volumes of data without sacrificing interpretability. The SEQC model is that of density-functional-based tight binding (DFTB) with fixed atomic orbital energies and interactions that are one-dimensional functions of the interatomic distance. This model is trained to ab initio data in a manner that is analogous to that used to train deep learning models. Using benchmarks that reflect the accuracy of the training data, we show that the resulting model maintains a physically reasonable functional form while achieving an accuracy, relative to coupled cluster energies with a complete basis set extrapolation (CCSD(T)*/CBS), that is comparable to that of density functional theory (DFT). This suggests that trained SEQC models can achieve a low computational cost and high accuracy without sacrificing interpretability. Use of a physically motivated model form also substantially reduces the amount of ab initio data needed to train the model compared to that required for deep learning models.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
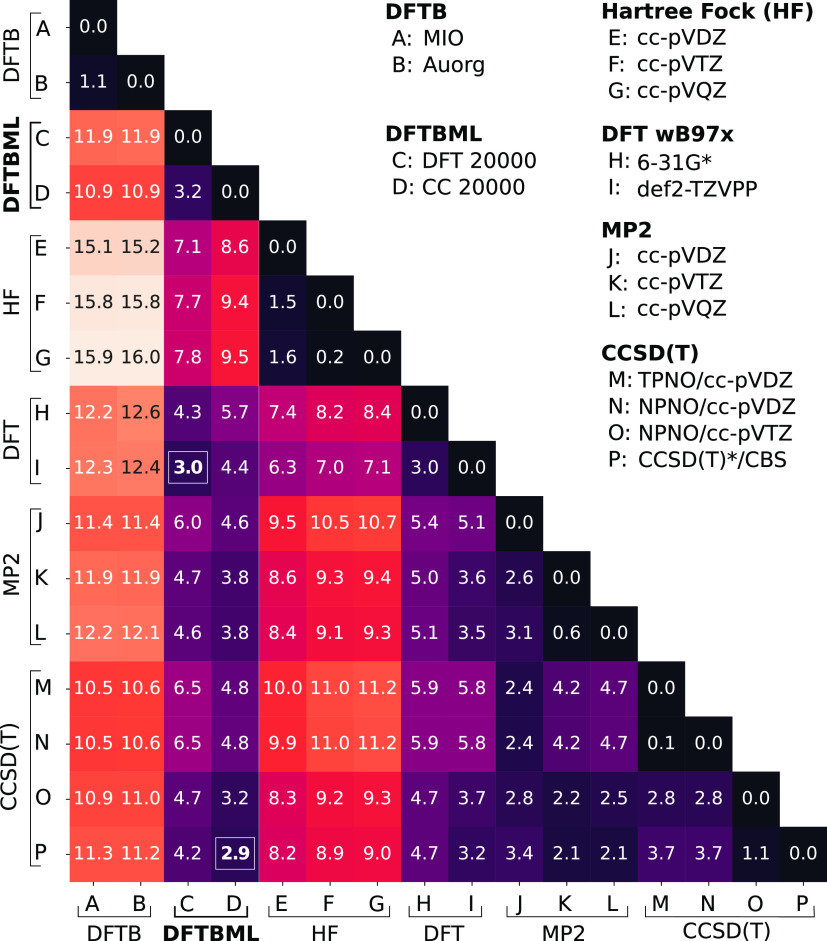
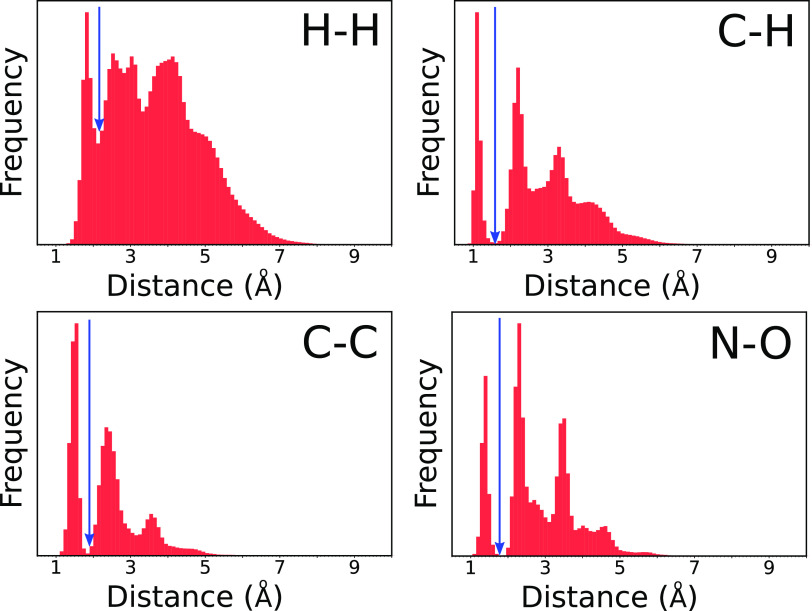

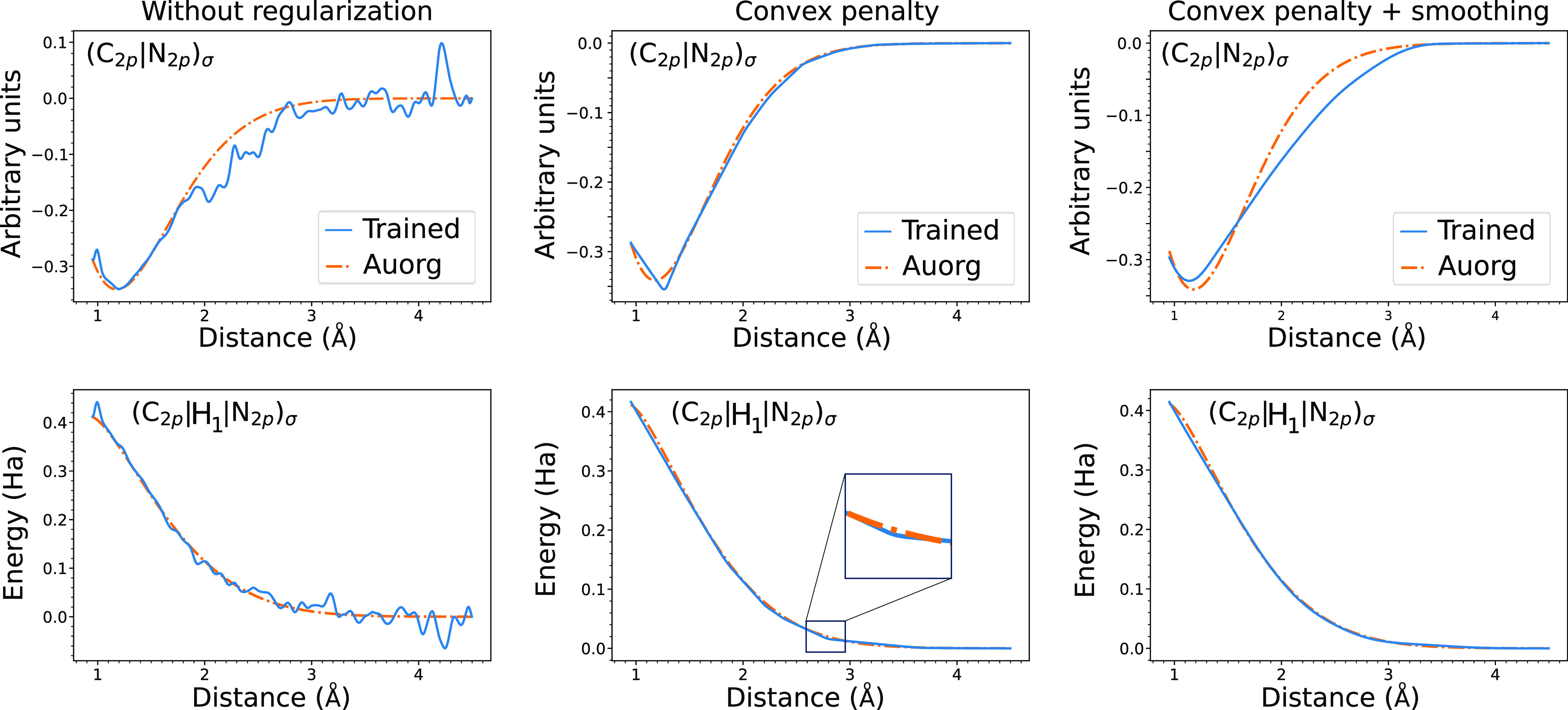
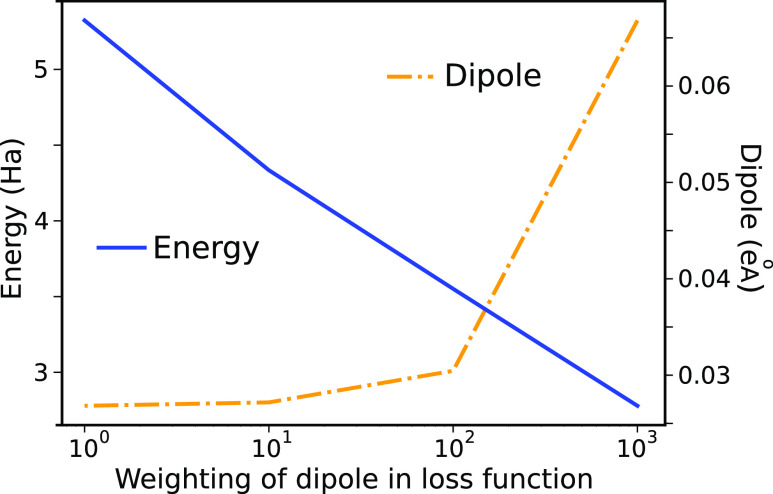
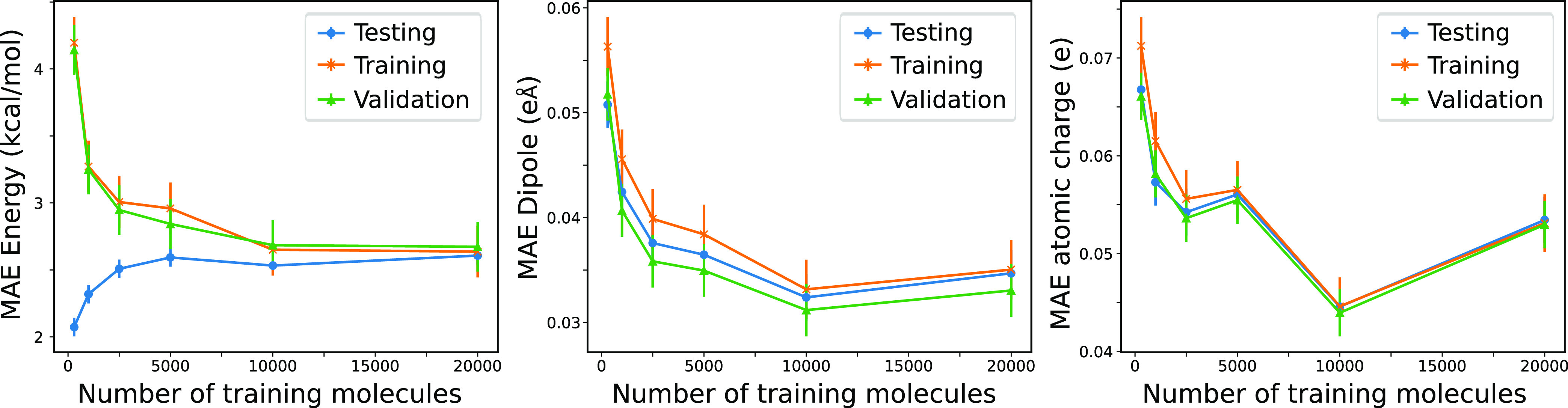
 where σ is the standard deviation
of the errors calculated separately for the training, validation,
and testing values.
where σ is the standard deviation
of the errors calculated separately for the training, validation,
and testing values.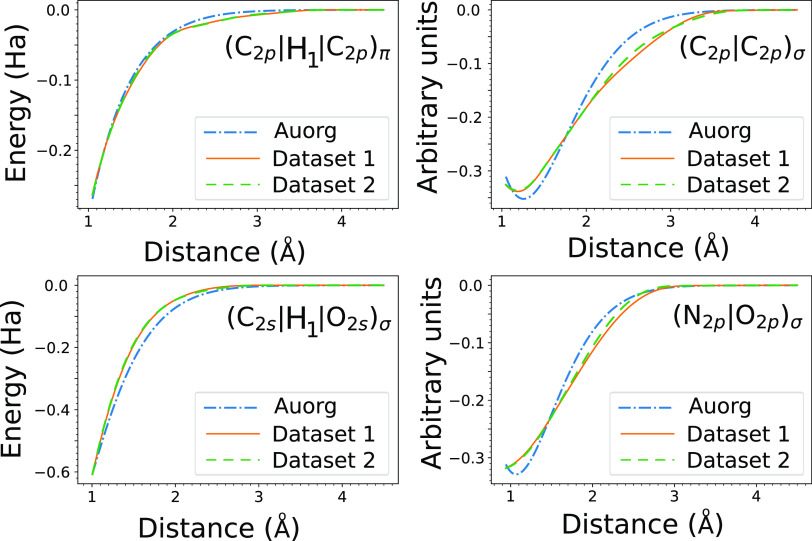
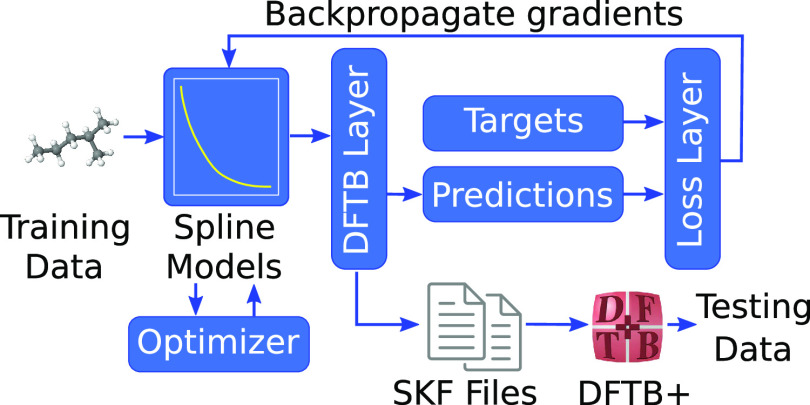
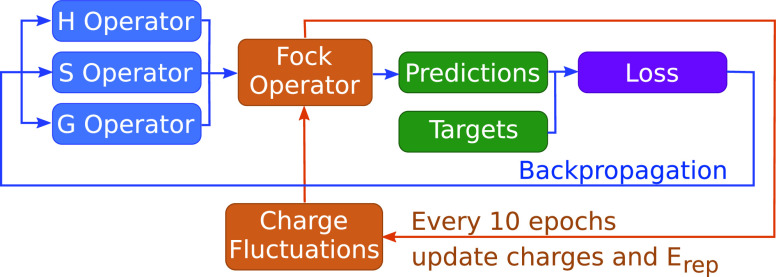
References
-
- Scuseria G. E. Comparison of coupled-cluster results with a hybrid of Hartree-Fock and density functional theory. J. Chem. Phys. 1992, 97, 7528–7530. 10.1063/1.463977. - DOI
-
- Gruber T.; Liao K.; Tsatsoulis T.; Hummel F.; Grüneis A. Applying the Coupled-Cluster Ansatz to Solids and Surfaces in the Thermodynamic Limit. Phys. Rev. X 2018, 8, 021043 10.1103/PhysRevX.8.021043. - DOI
LinkOut - more resources
Full Text Sources
