

Metabolic targeting of platelets to combat thrombosis: dawn of a new paradigm?
- PMID: 37706546
- PMCID: PMC10676458
- DOI: 10.1093/cvr/cvad149
Metabolic targeting of platelets to combat thrombosis: dawn of a new paradigm?
Abstract
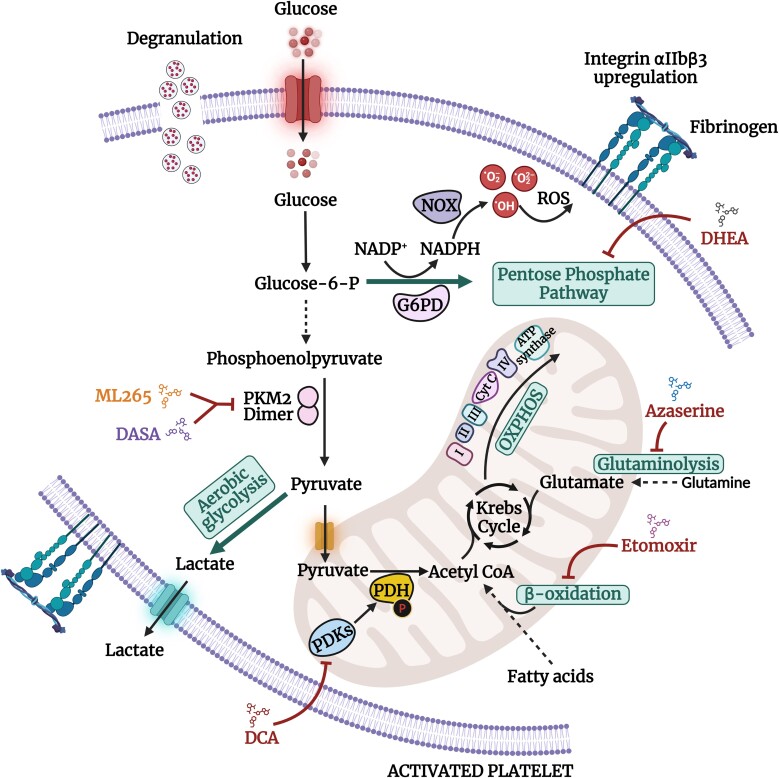
Current antithrombotic therapies used in clinical settings target either the coagulation pathways or platelet activation receptors (P2Y12 or GPIIb/IIIa), as well as the cyclooxygenase (COX) enzyme through aspirin. However, they are associated with bleeding risk and are not suitable for long-term use. Thus, novel strategies which provide broad protection against platelet activation with minimal bleeding risks are required. Regardless of the nature of agonist stimulation, platelet activation is an energy-intensive and ATP-driven process characterized by metabolic switching toward a high rate of aerobic glycolysis, relative to oxidative phosphorylation (OXPHOS). Consequently, there has been considerable interest in recent years in investigating whether targeting metabolic pathways in platelets, especially aerobic glycolysis and OXPHOS, can modulate their activation, thereby preventing thrombosis. This review briefly discusses the choices of metabolic substrates available to platelets that drive their metabolic flexibility. We have comprehensively elucidated the relevance of aerobic glycolysis in facilitating platelet activation and the underlying molecular mechanisms that trigger this switch from OXPHOS. We have provided a detailed account of the antiplatelet effects of targeting vital metabolic checkpoints such as pyruvate dehydrogenase kinases (PDKs) and pyruvate kinase M2 (PKM2) that preferentially drive the pyruvate flux to aerobic glycolysis. Furthermore, we discuss the role of fatty acids and glutamine oxidation in mitochondria and their subsequent role in driving OXPHOS and platelet activation. While the approach of targeting metabolic regulatory mechanisms in platelets to prevent their activation is still in a nascent stage, accumulating evidence highlights its beneficial effects as a potentially novel antithrombotic strategy.
Keywords: Platelets • Aerobic glycolysis • OXPHOS • Thrombosis.
© The Author(s) 2023. Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
Conflict of interest: none declared.
Figures



References
-
- Tatsumi K, Mackman N. Tissue factor and atherothrombosis. J Atheroscler Thromb 2015;22:543–549. - PubMed
-
- van Uden R, Houtenbos I, Griffioen-Keijzer A, Odekerken DAM, van den Bemt P, Becker ML. Guidelines for mono, double and triple antithrombotic therapy. Postgrad Med J 2021;97:730–737. - PubMed
-
- Aibibula M, Naseem KM, Sturmey RG. Glucose metabolism and metabolic flexibility in blood platelets. J Thromb Haemost 2018;16:2300–2314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
