

MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer's disease
- PMID: 37708272
- PMCID: PMC7615236
- DOI: 10.1126/science.abp9556
MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer's disease
Abstract
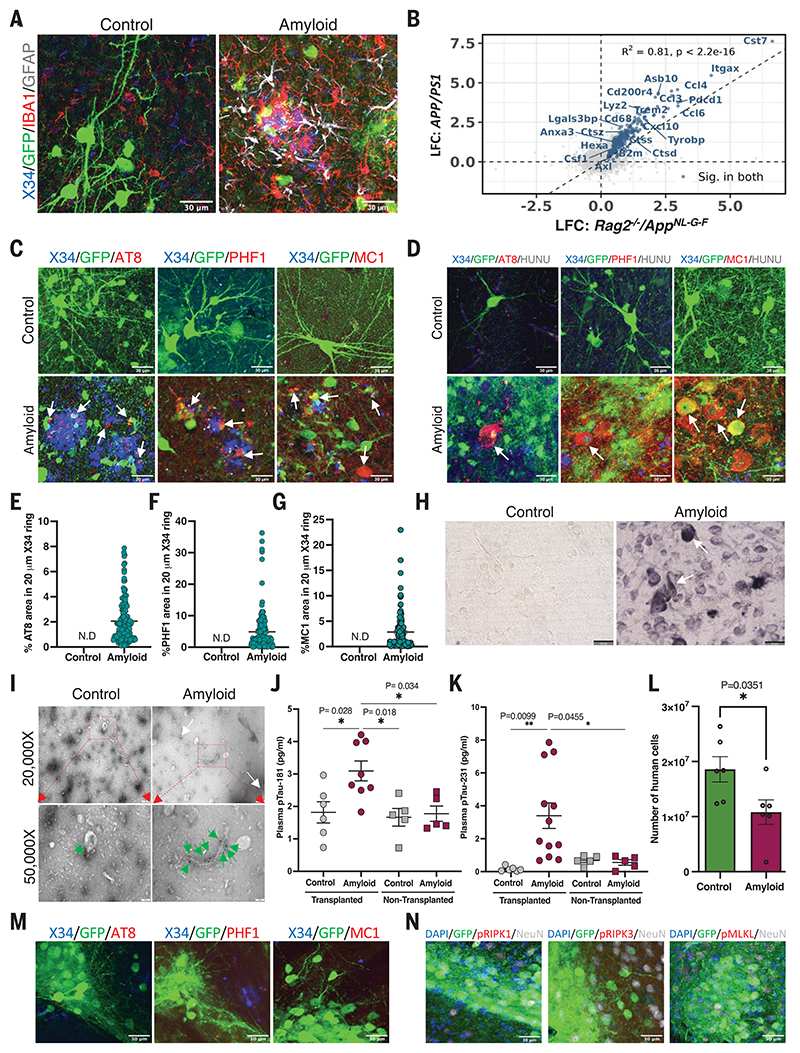
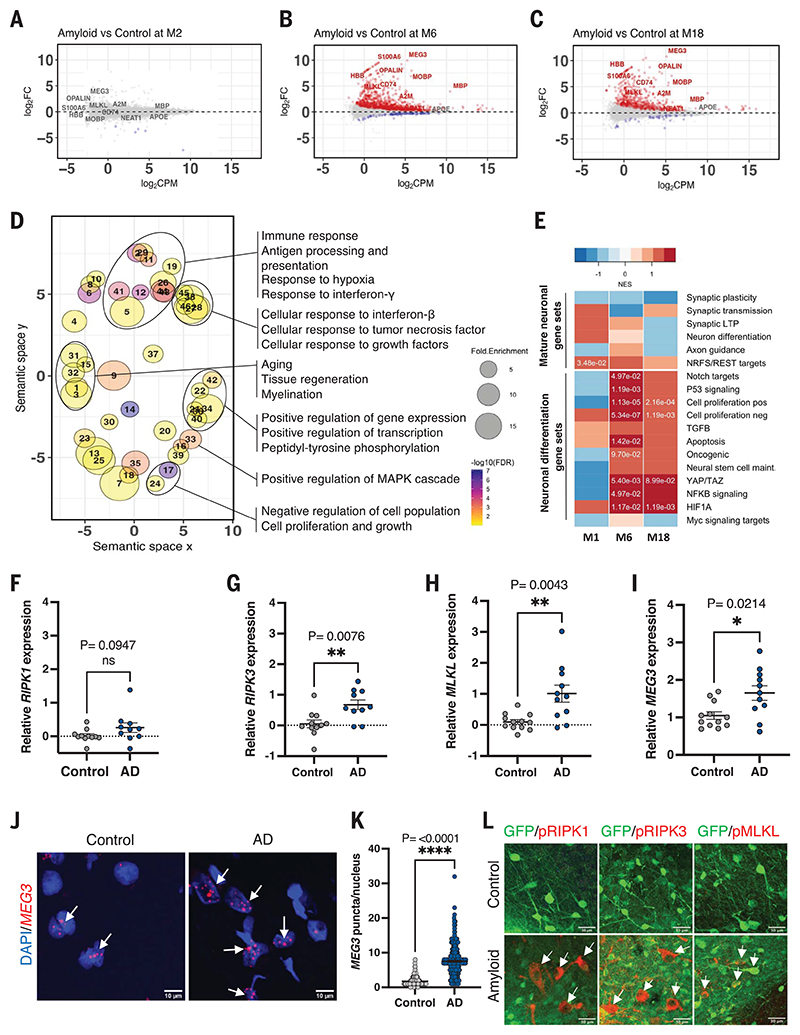
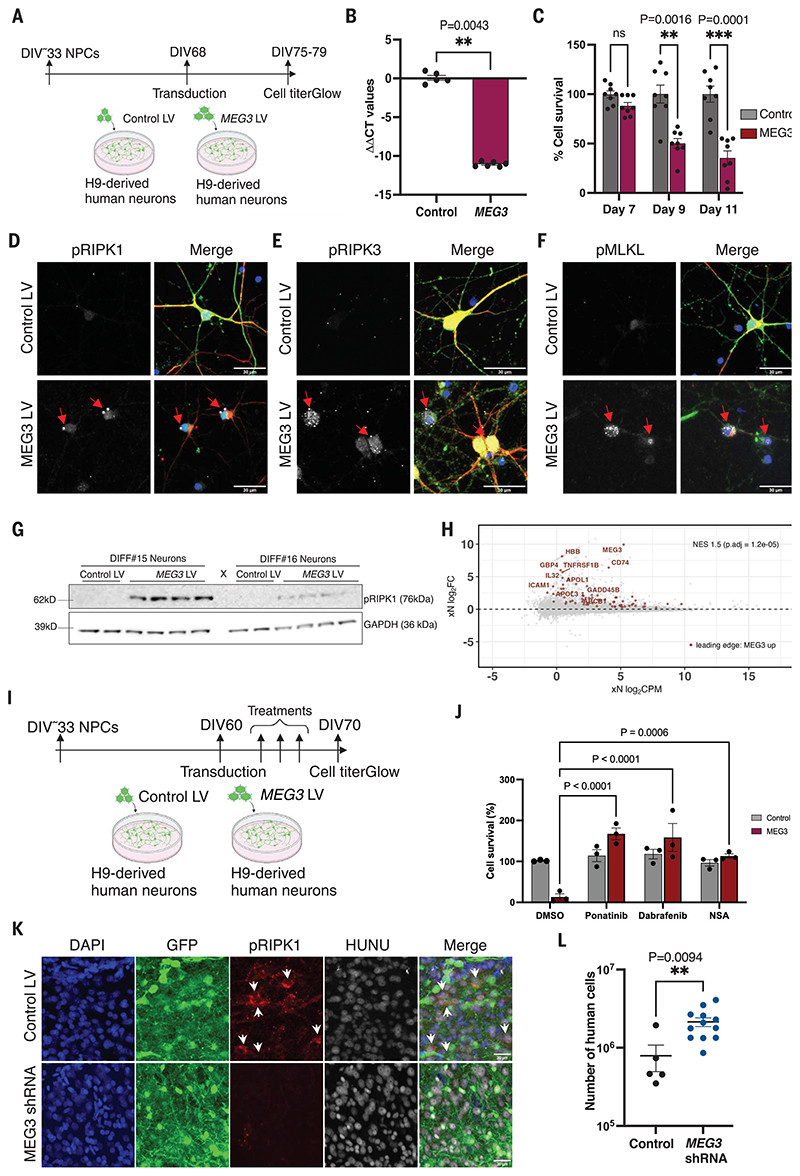
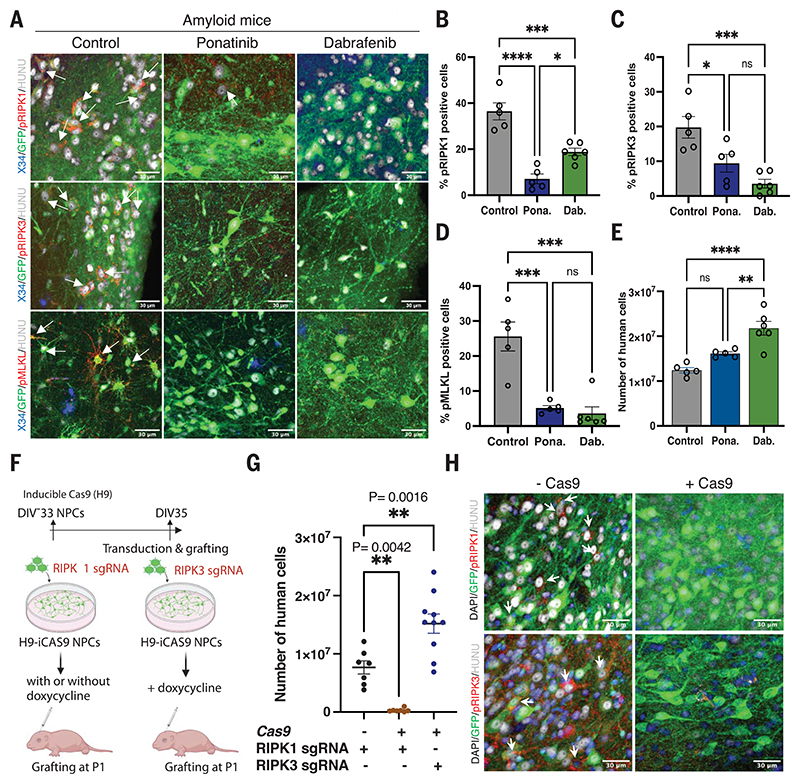
Neuronal cell loss is a defining feature of Alzheimer's disease (AD), but the underlying mechanisms remain unclear. We xenografted human or mouse neurons into the brain of a mouse model of AD. Only human neurons displayed tangles, Gallyas silver staining, granulovacuolar neurodegeneration (GVD), phosphorylated tau blood biomarkers, and considerable neuronal cell loss. The long noncoding RNA MEG3 was strongly up-regulated in human neurons. This neuron-specific long noncoding RNA is also up-regulated in AD patients. MEG3 expression alone was sufficient to induce necroptosis in human neurons in vitro. Down-regulation of MEG3 and inhibition of necroptosis using pharmacological or genetic manipulation of receptor-interacting protein kinase 1 (RIPK1), RIPK3, or mixed lineage kinase domain-like protein (MLKL) rescued neuronal cell loss in xenografted human neurons. This model suggests potential therapeutic approaches for AD and reveals a human-specific vulnerability to AD.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
