

Endoplasmic reticulum stress: molecular mechanism and therapeutic targets
- PMID: 37709773
- PMCID: PMC10502142
- DOI: 10.1038/s41392-023-01570-w
Endoplasmic reticulum stress: molecular mechanism and therapeutic targets
Abstract
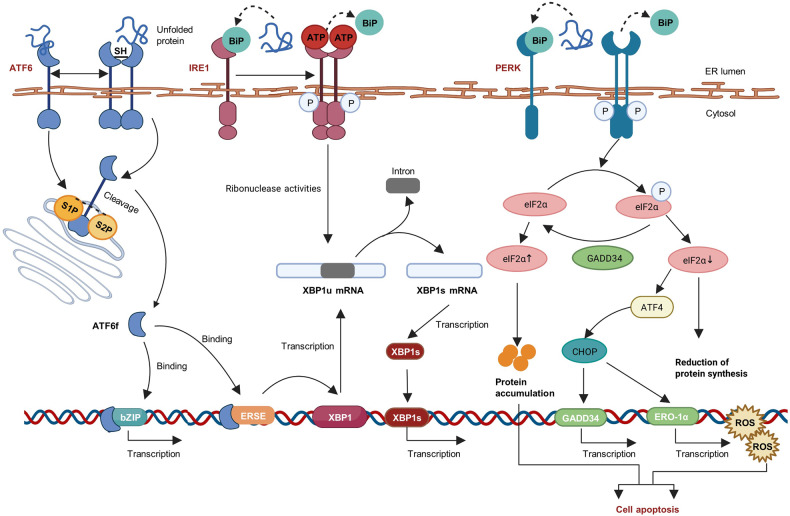
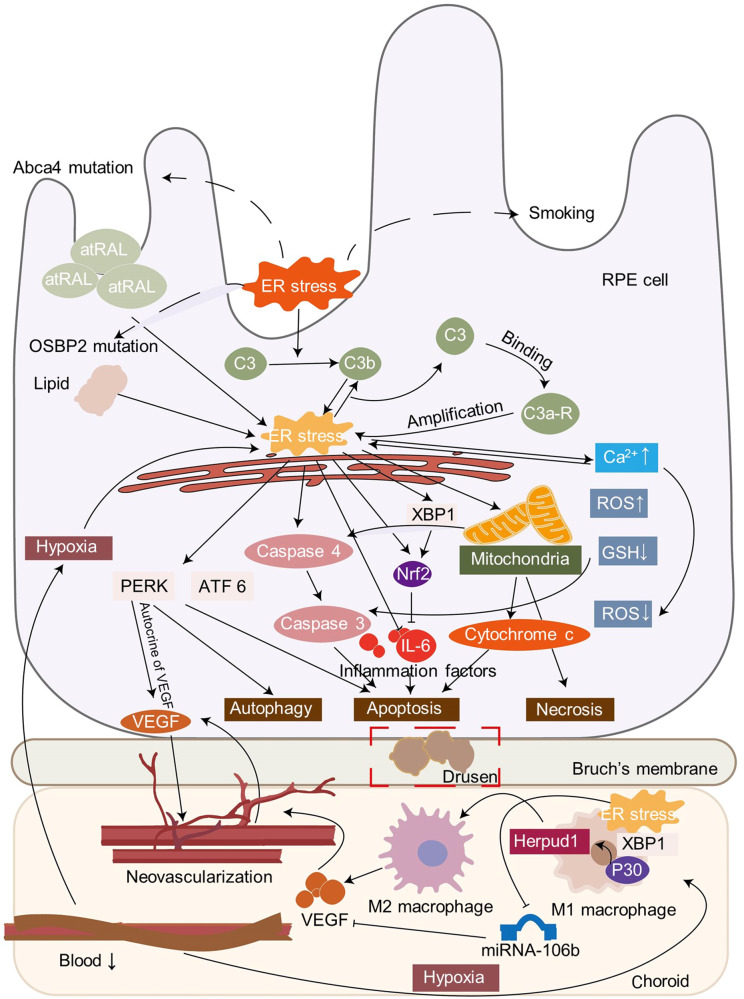
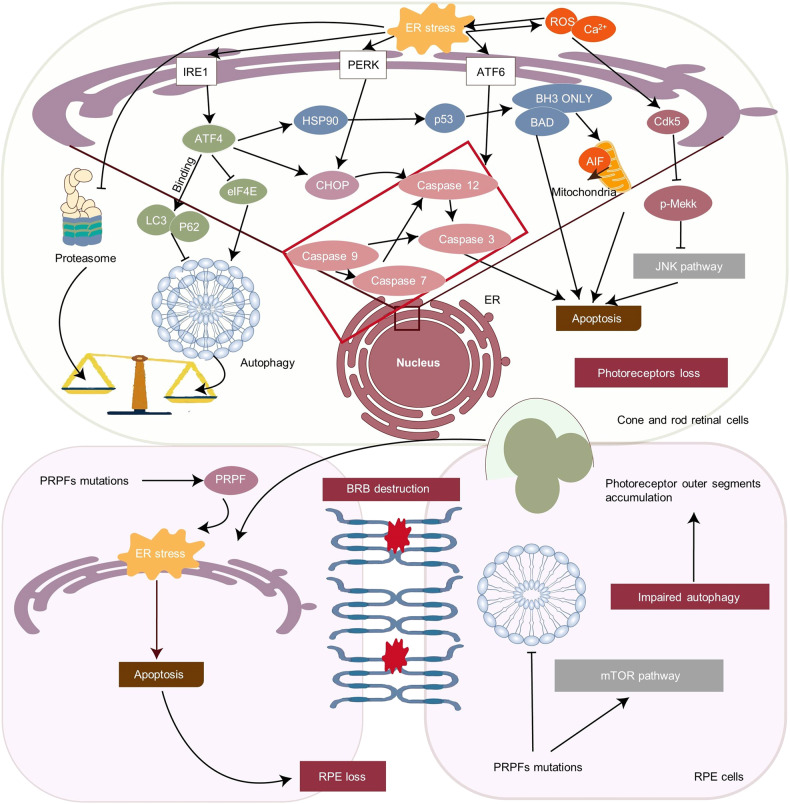
The endoplasmic reticulum (ER) functions as a quality-control organelle for protein homeostasis, or "proteostasis". The protein quality control systems involve ER-associated degradation, protein chaperons, and autophagy. ER stress is activated when proteostasis is broken with an accumulation of misfolded and unfolded proteins in the ER. ER stress activates an adaptive unfolded protein response to restore proteostasis by initiating protein kinase R-like ER kinase, activating transcription factor 6, and inositol requiring enzyme 1. ER stress is multifaceted, and acts on aspects at the epigenetic level, including transcription and protein processing. Accumulated data indicates its key role in protein homeostasis and other diverse functions involved in various ocular diseases, such as glaucoma, diabetic retinopathy, age-related macular degeneration, retinitis pigmentosa, achromatopsia, cataracts, ocular tumors, ocular surface diseases, and myopia. This review summarizes the molecular mechanisms underlying the aforementioned ocular diseases from an ER stress perspective. Drugs (chemicals, neurotrophic factors, and nanoparticles), gene therapy, and stem cell therapy are used to treat ocular diseases by alleviating ER stress. We delineate the advancement of therapy targeting ER stress to provide new treatment strategies for ocular diseases.
© 2023. West China Hospital, Sichuan University.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. - PubMed
-
- Kaushik S, Cuervo AM. Proteostasis and aging. Nat. Med. 2015;21:1406–1415. - PubMed
-
- Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
