

Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation
- PMID: 37710018
- PMCID: PMC11222905
- DOI: 10.1038/s41592-023-01993-x
Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation
Abstract
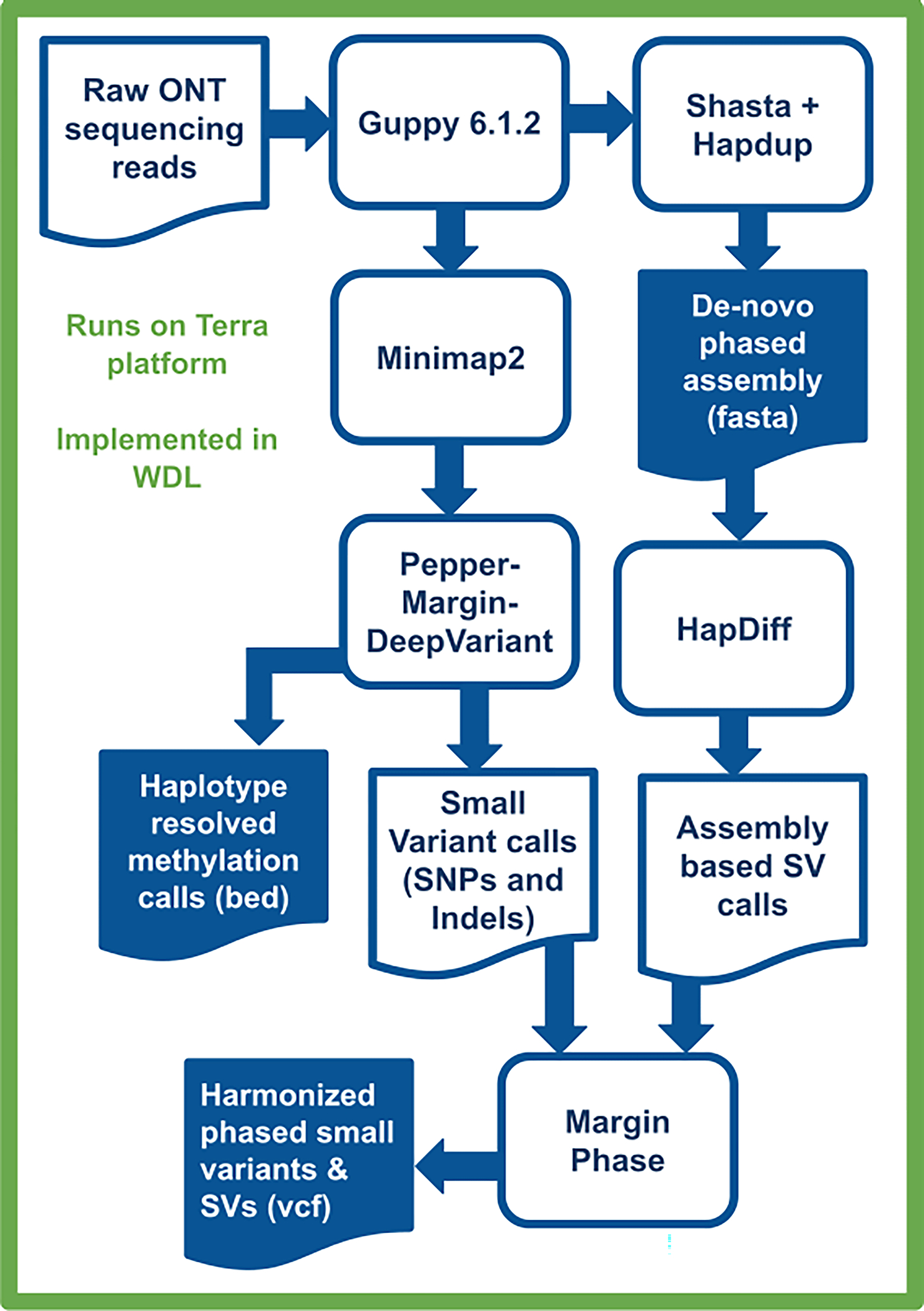
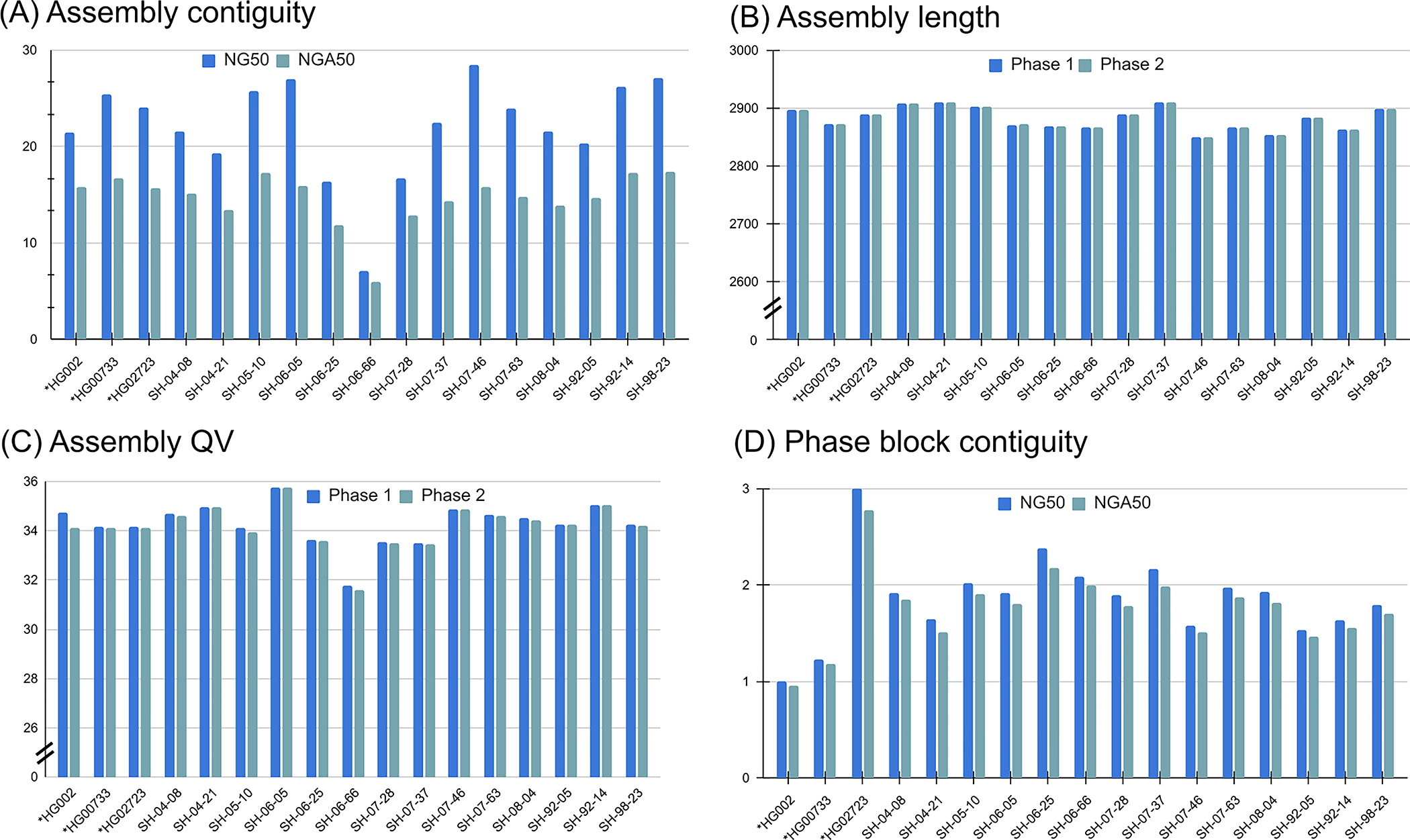
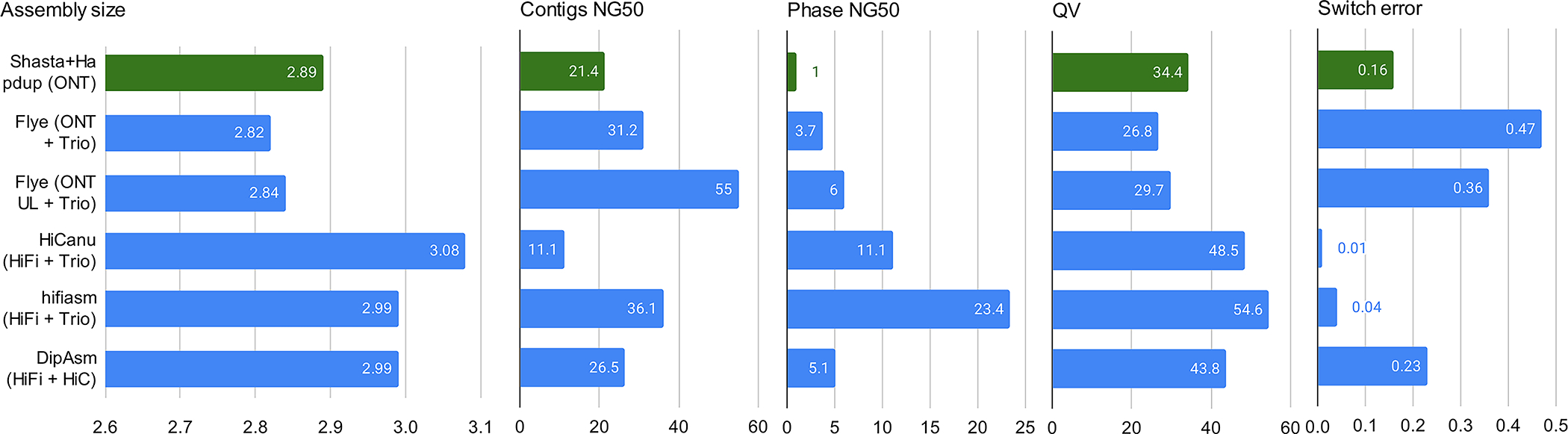
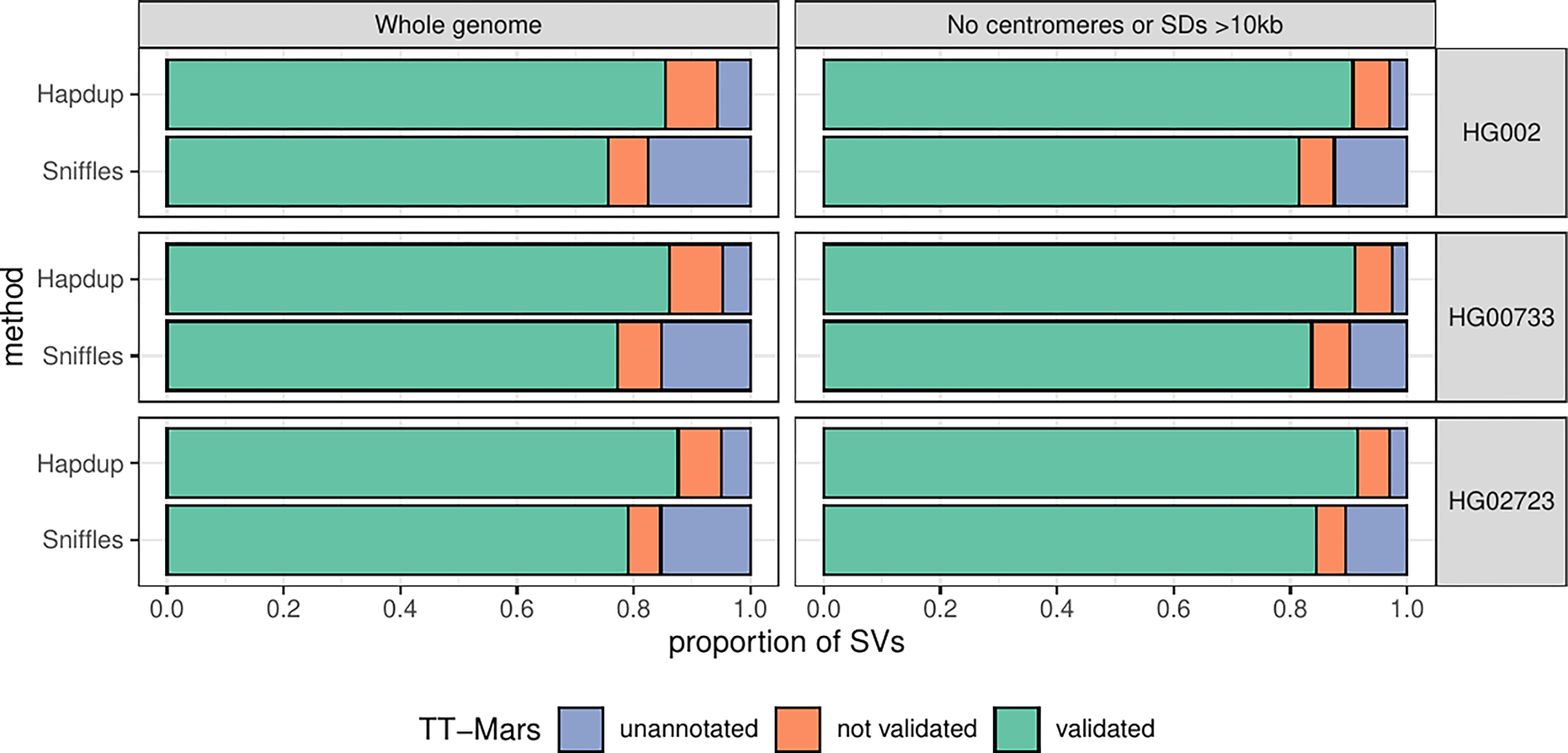
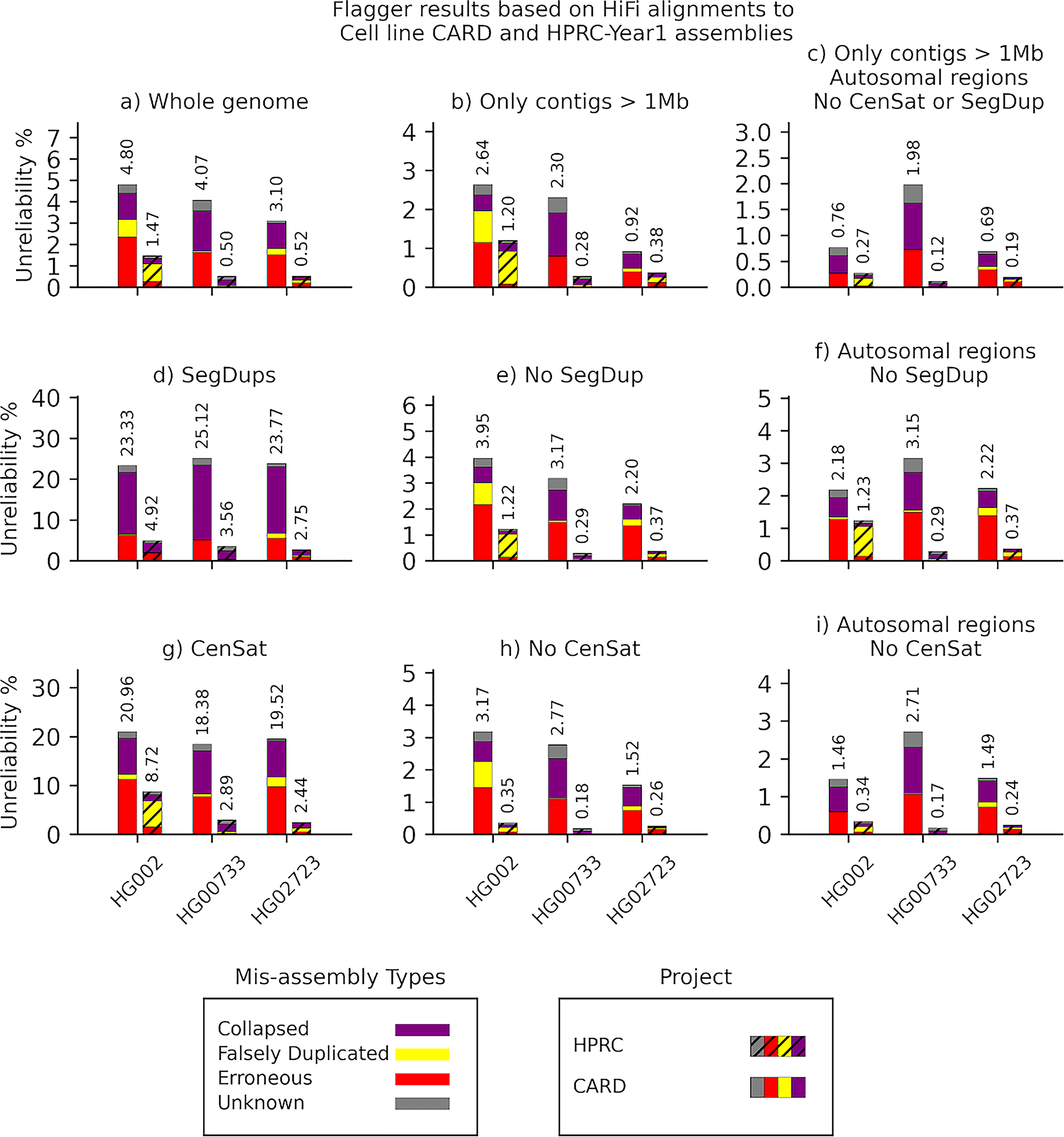
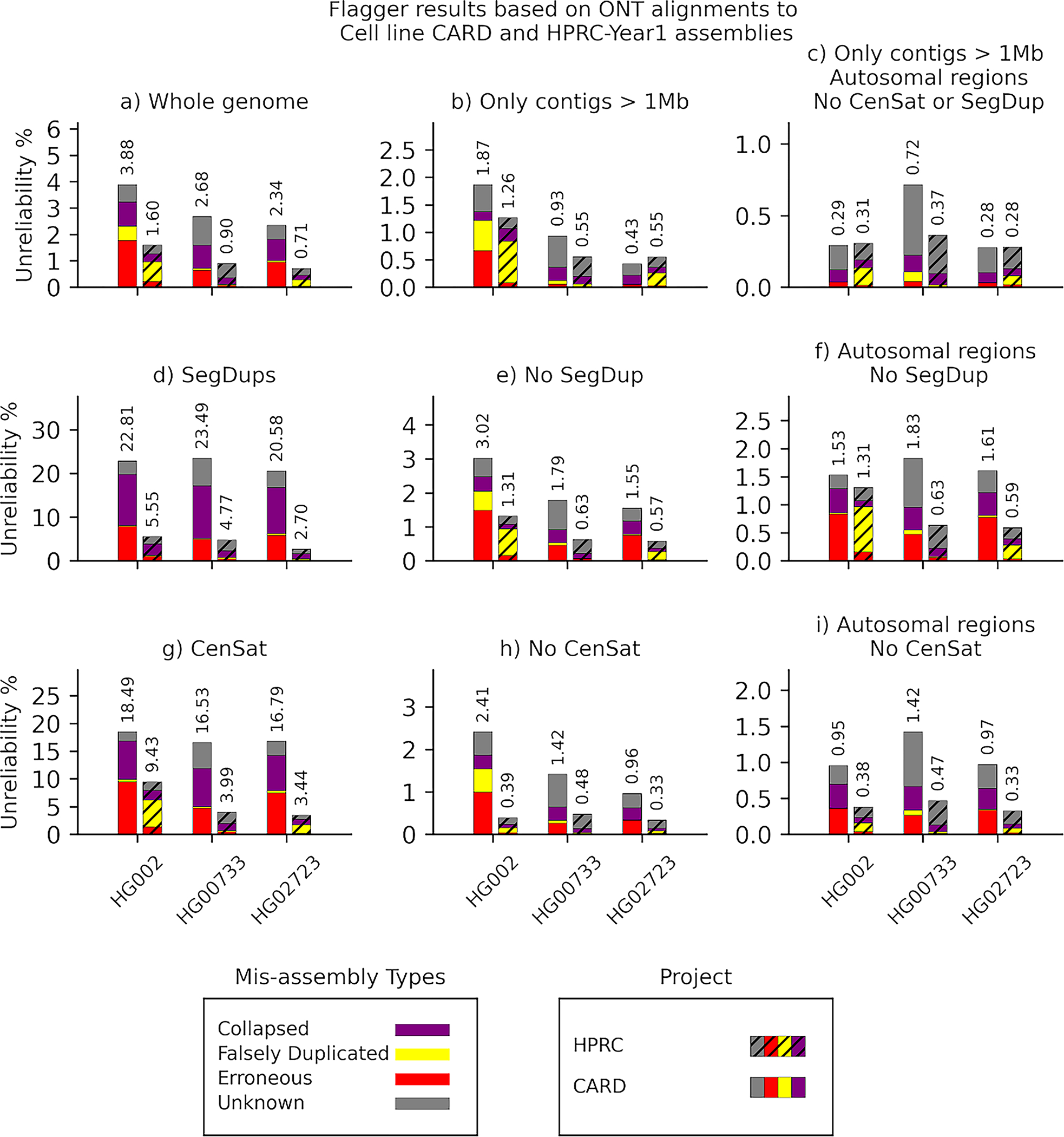
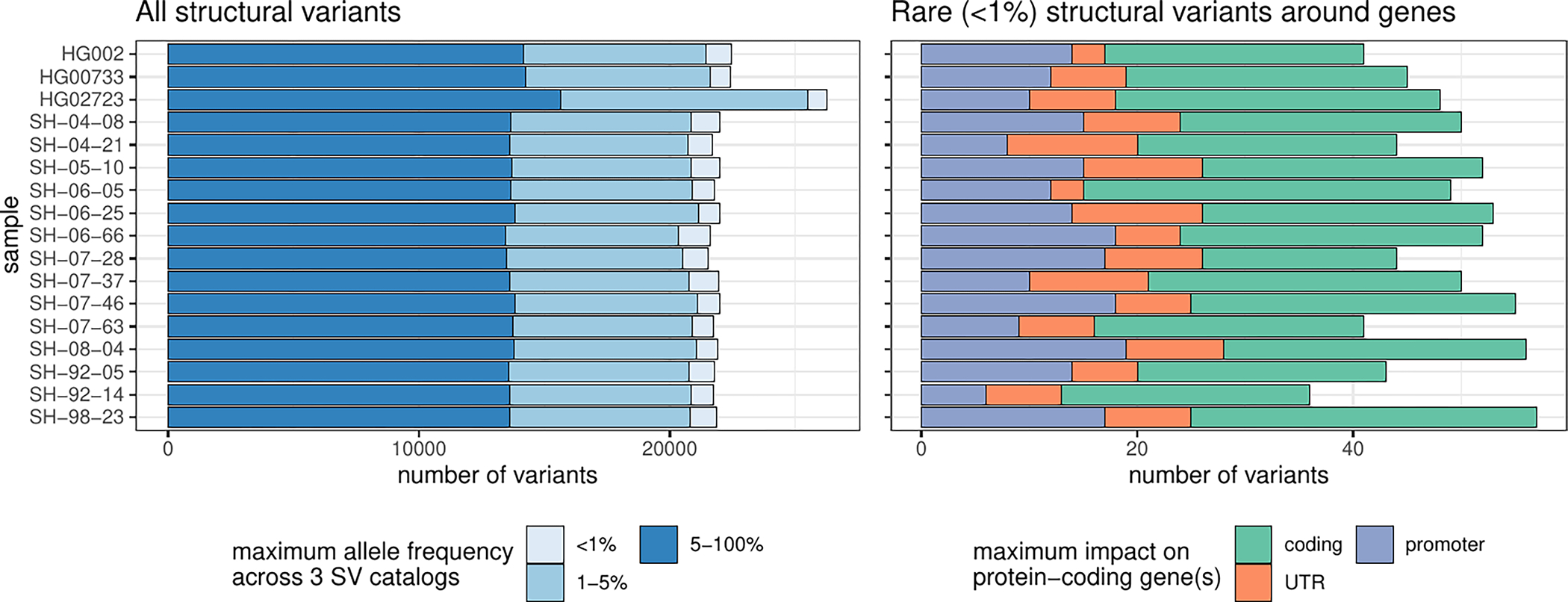
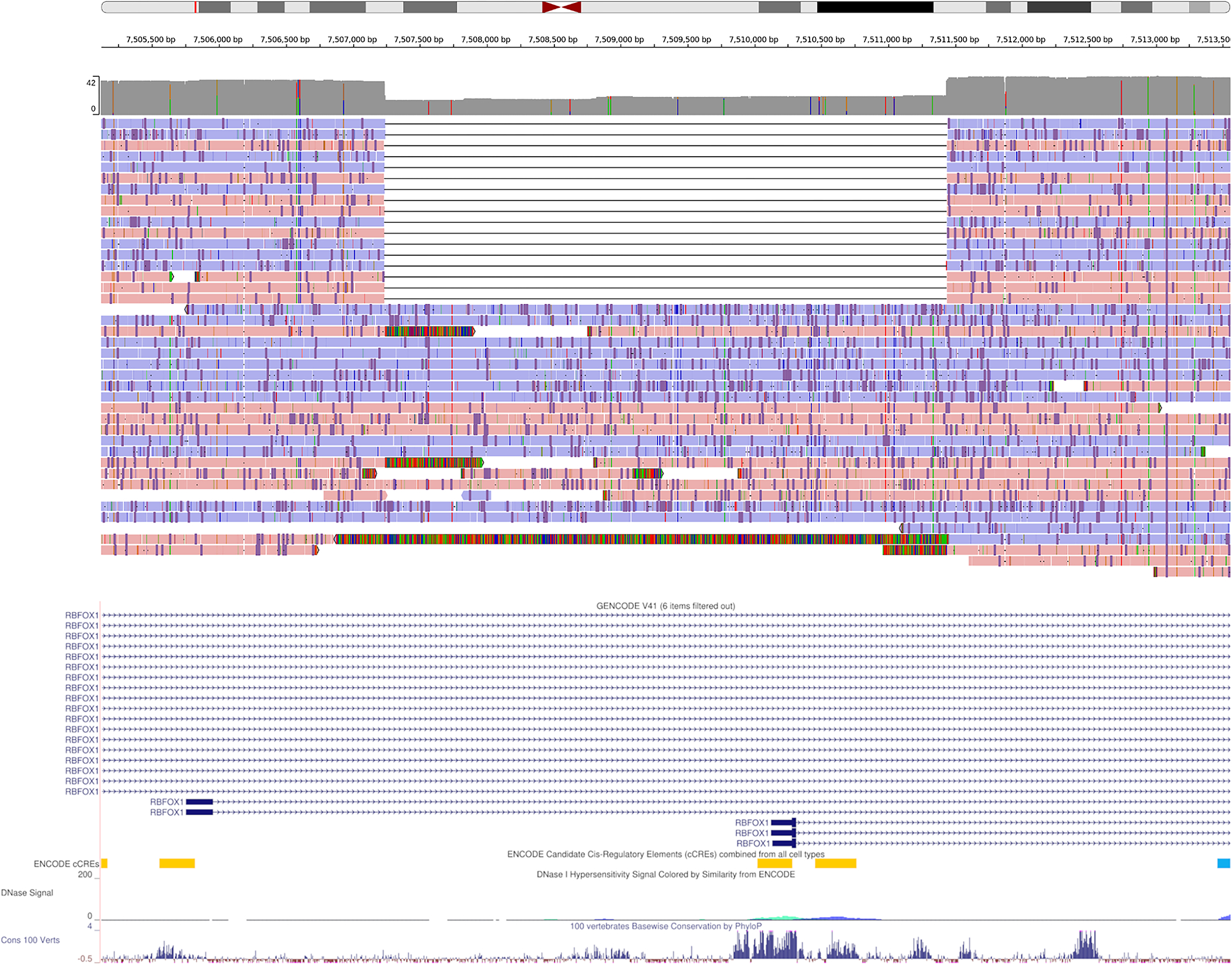
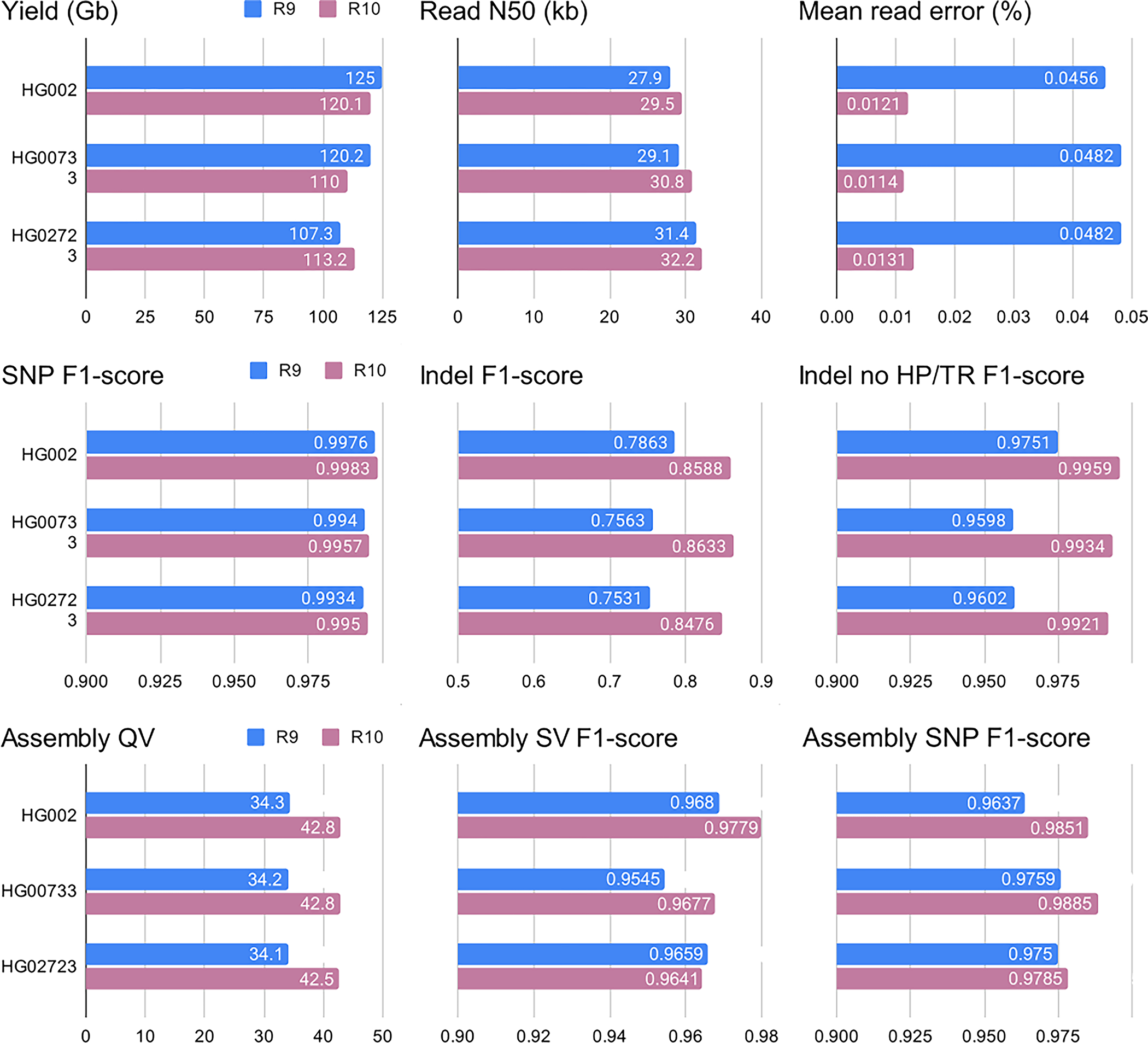
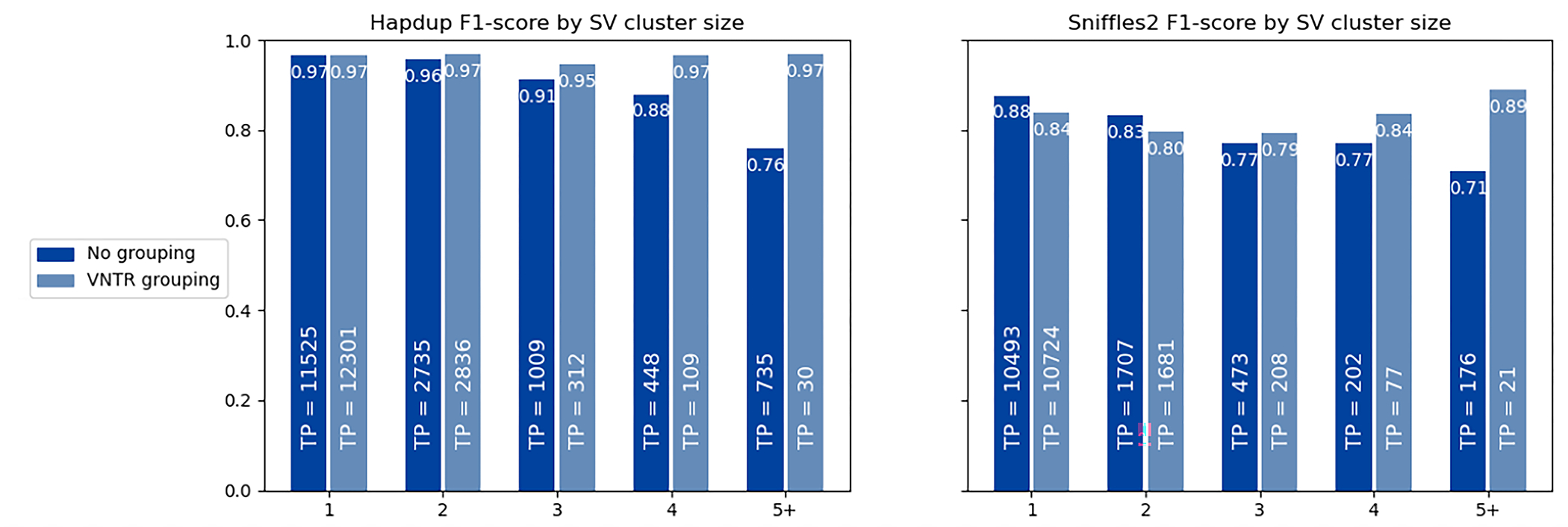
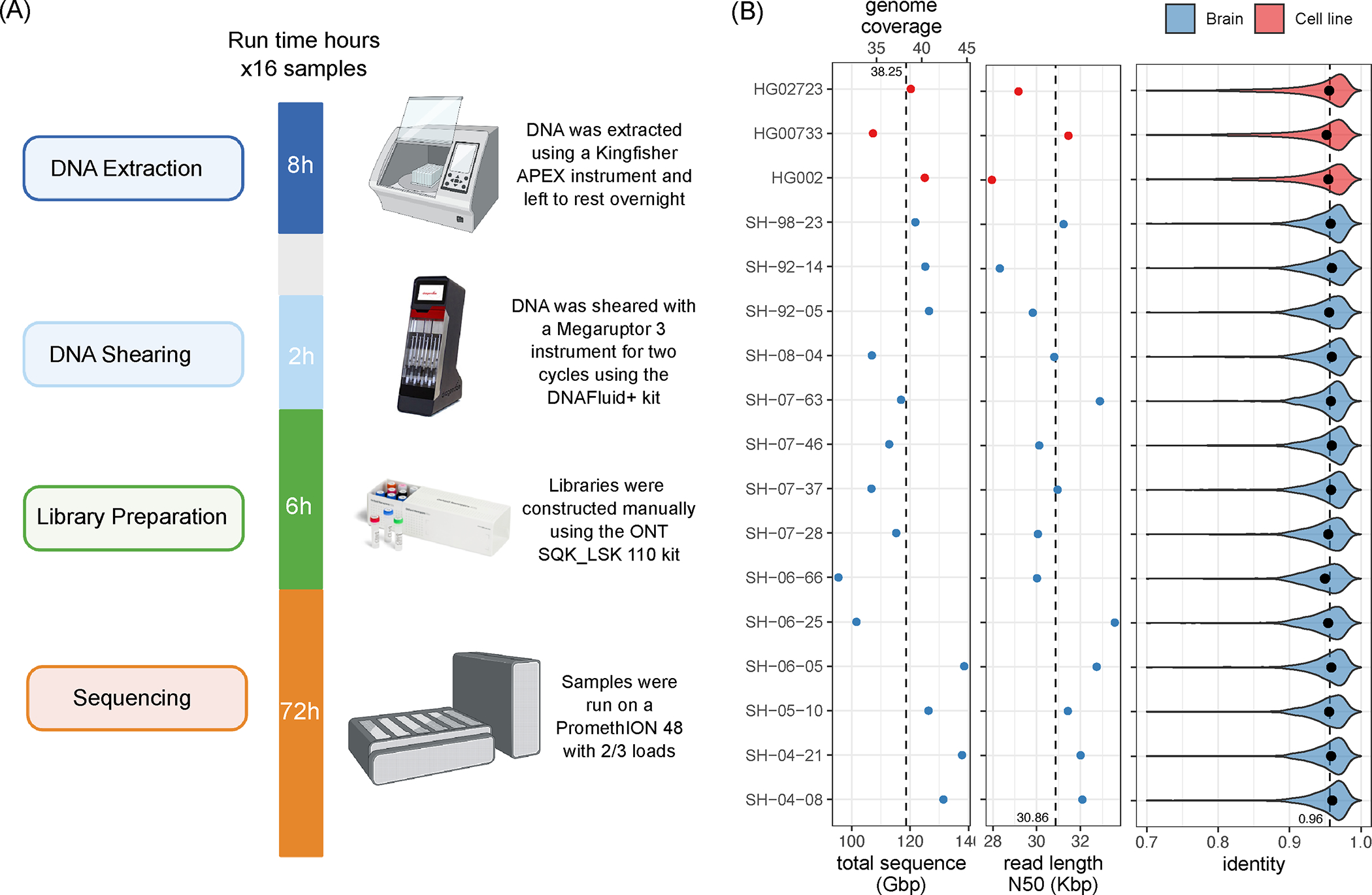
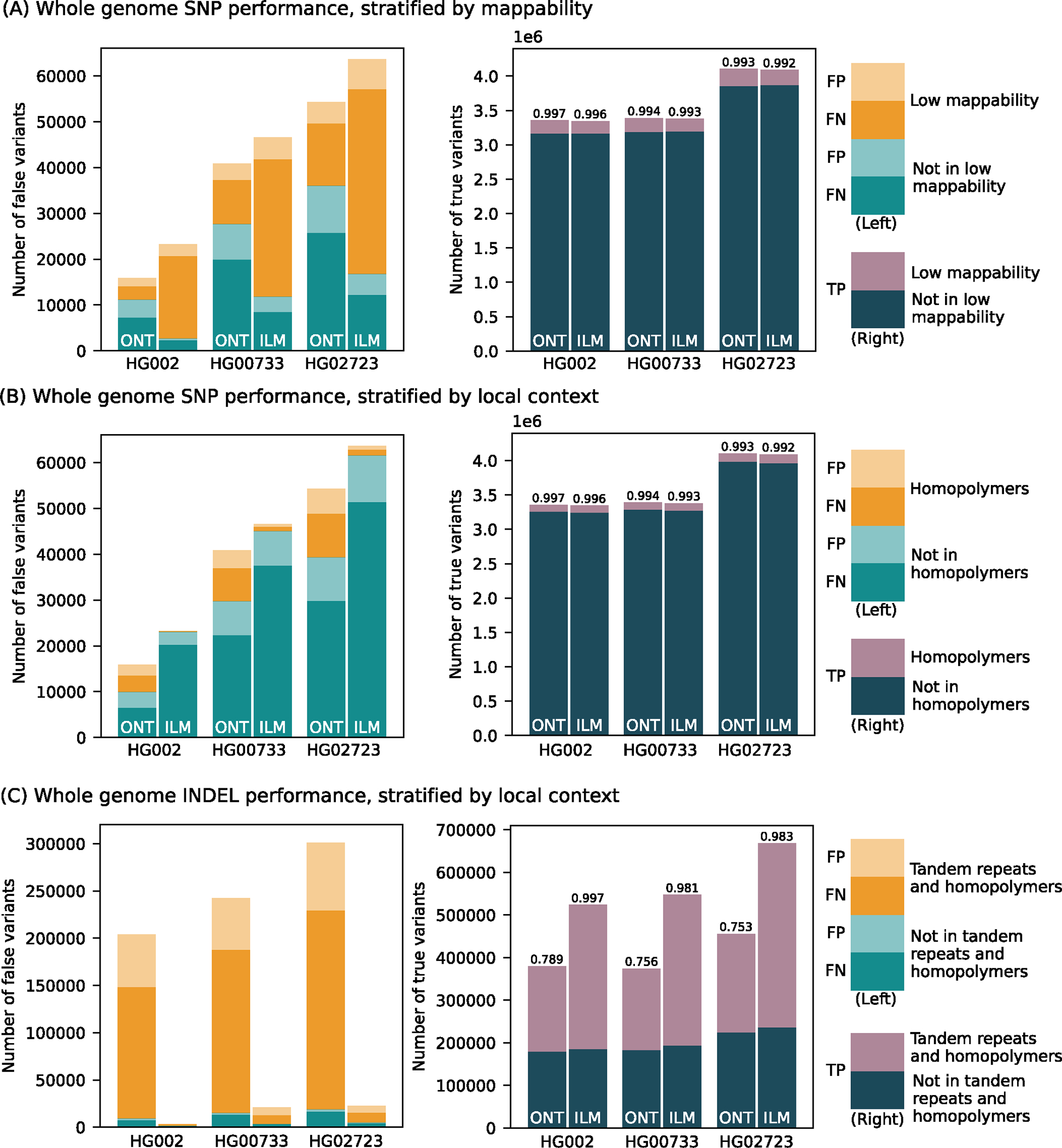
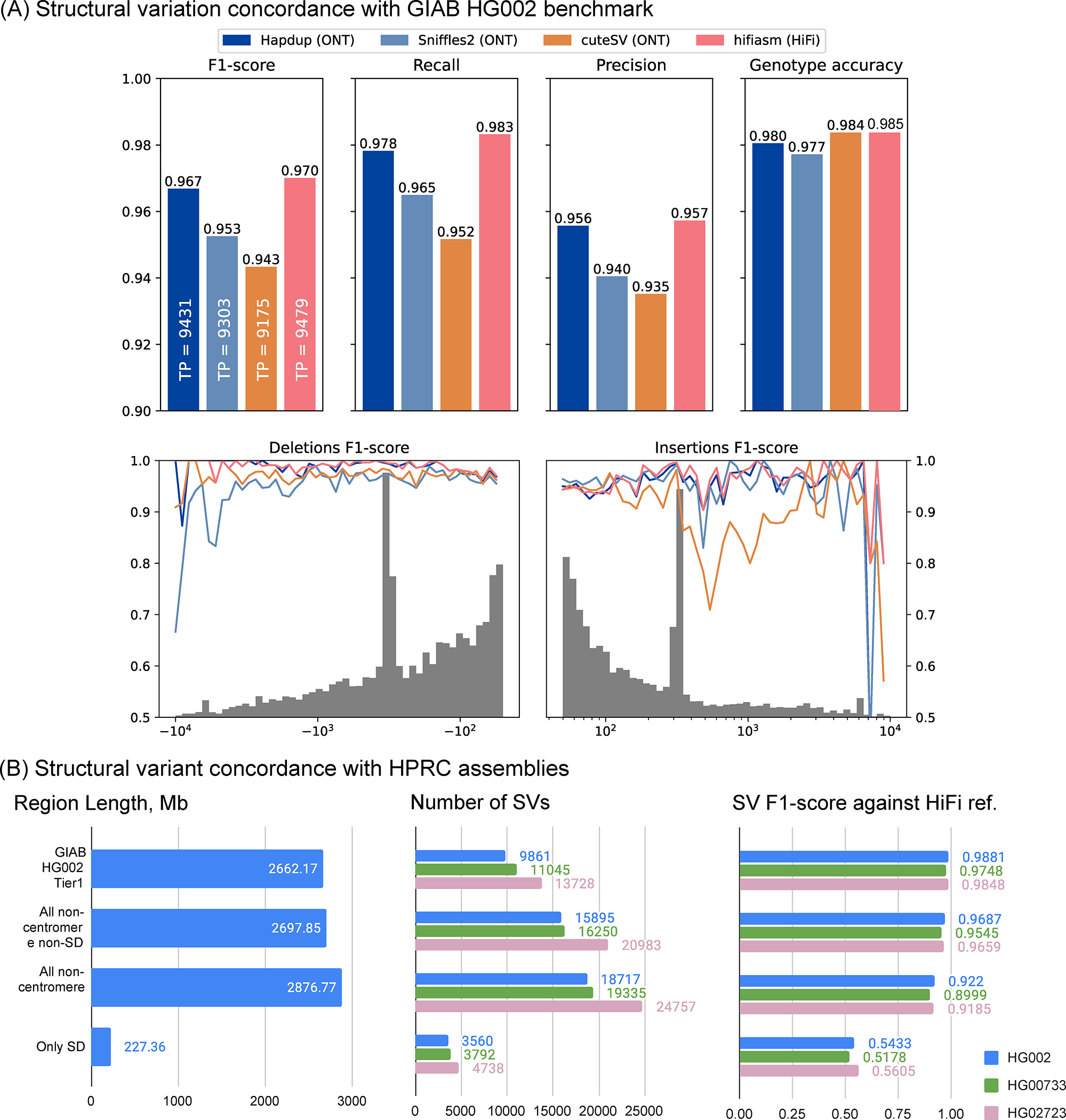
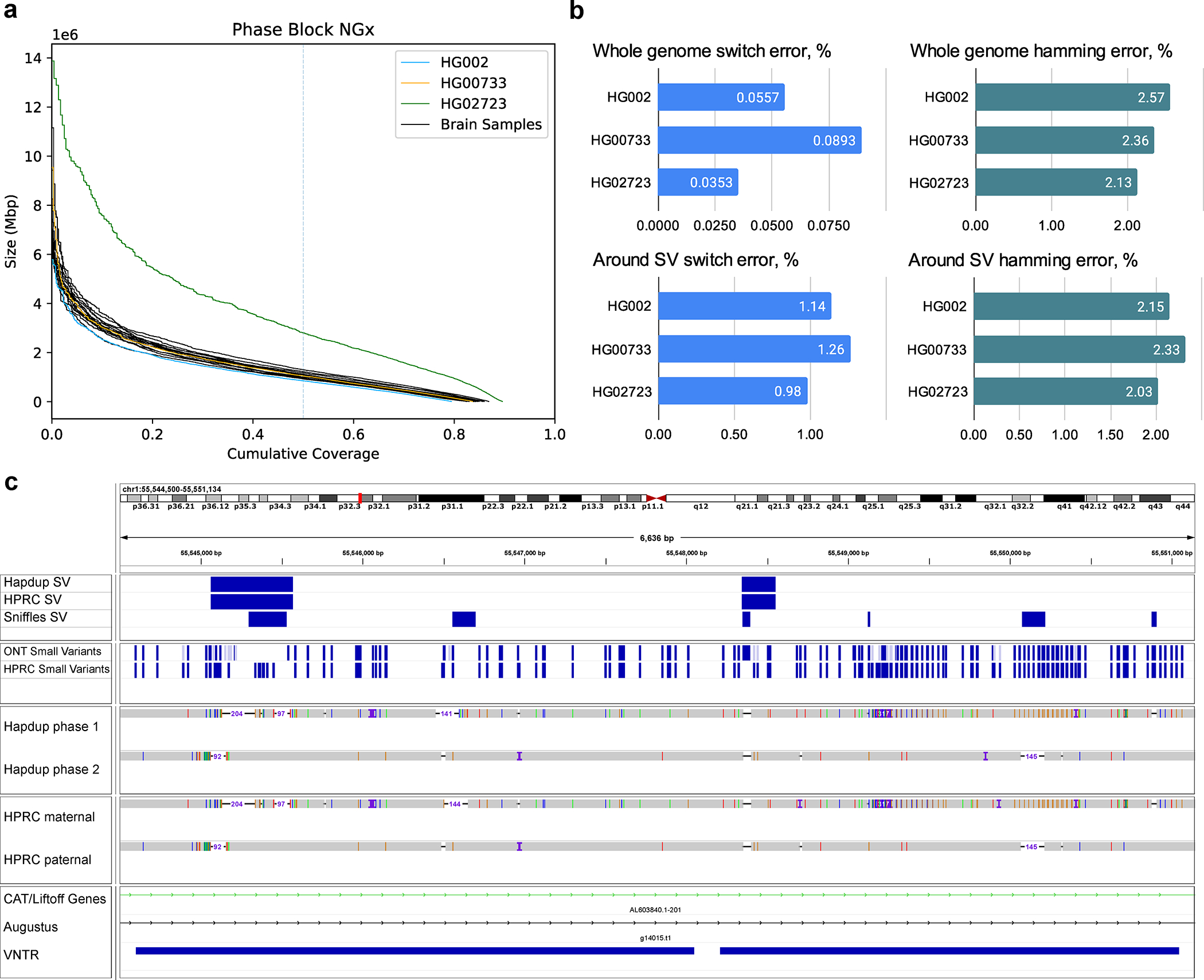
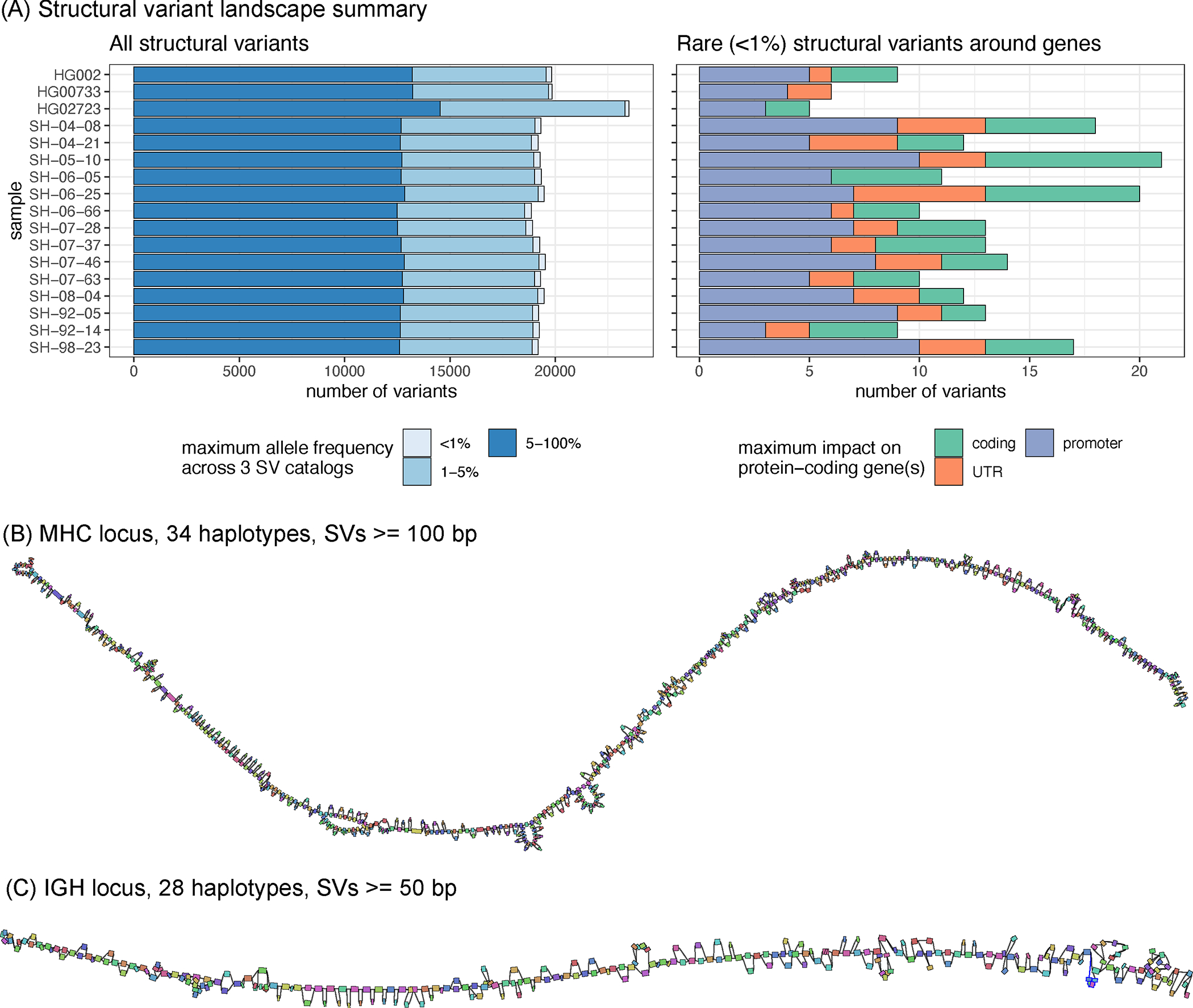
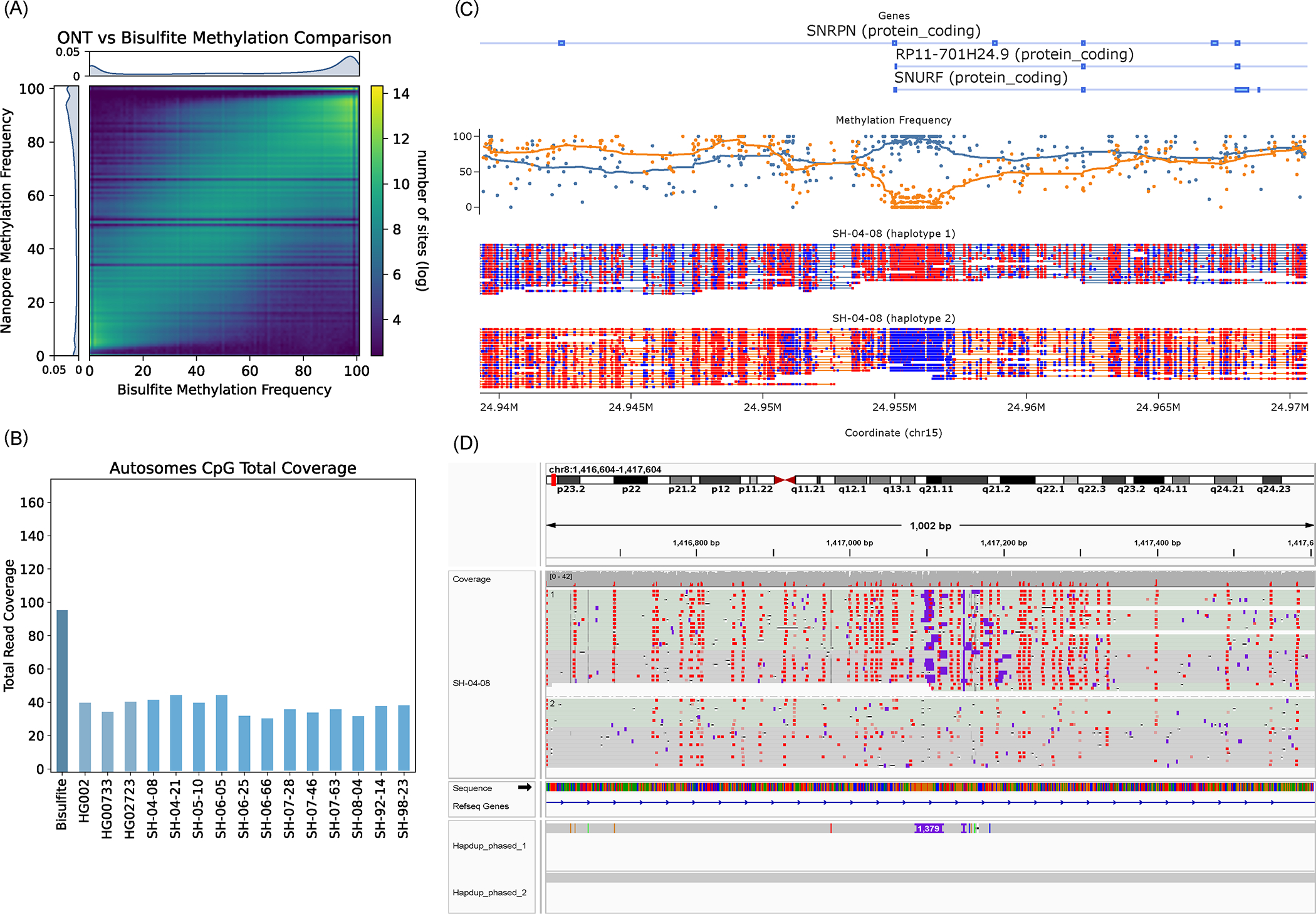
Long-read sequencing technologies substantially overcome the limitations of short-reads but have not been considered as a feasible replacement for population-scale projects, being a combination of too expensive, not scalable enough or too error-prone. Here we develop an efficient and scalable wet lab and computational protocol, Napu, for Oxford Nanopore Technologies long-read sequencing that seeks to address those limitations. We applied our protocol to cell lines and brain tissue samples as part of a pilot project for the National Institutes of Health Center for Alzheimer's and Related Dementias. Using a single PromethION flow cell, we can detect single nucleotide polymorphisms with F1-score comparable to Illumina short-read sequencing. Small indel calling remains difficult within homopolymers and tandem repeats, but achieves good concordance to Illumina indel calls elsewhere. Further, we can discover structural variants with F1-score on par with state-of-the-art de novo assembly methods. Our protocol phases small and structural variants at megabase scales and produces highly accurate, haplotype-specific methylation calls.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Figures
Update of
-
Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation.bioRxiv [Preprint]. 2023 Apr 5:2023.01.12.523790. doi: 10.1101/2023.01.12.523790. bioRxiv. 2023. Update in: Nat Methods. 2023 Oct;20(10):1483-1492. doi: 10.1038/s41592-023-01993-x. PMID: 36711673 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- T32 HG012344/HG/NHGRI NIH HHS/United States
- P01 AG000538/AG/NIA NIH HHS/United States
- P30 AG072980/AG/NIA NIH HHS/United States
- OT3 HL142481/HL/NHLBI NIH HHS/United States
- OT2 OD033761/OD/NIH HHS/United States
- U24 HG010262/HG/NHGRI NIH HHS/United States
- U24 HG011853/HG/NHGRI NIH HHS/United States
- P30 AG019610/AG/NIA NIH HHS/United States
- ZIA AG000538/ImNIH/Intramural NIH HHS/United States
- R01 HG009190/HG/NHGRI NIH HHS/United States
- OT2 OD026682/OD/NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- ZIA NS003154/ImNIH/Intramural NIH HHS/United States
- U24 NS072026/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
