

Clinical and molecular delineation of classical-like Ehlers-Danlos syndrome through a comprehensive next-generation sequencing-based screening system
- PMID: 37712068
- PMCID: PMC10498456
- DOI: 10.3389/fgene.2023.1234804
Clinical and molecular delineation of classical-like Ehlers-Danlos syndrome through a comprehensive next-generation sequencing-based screening system
Abstract
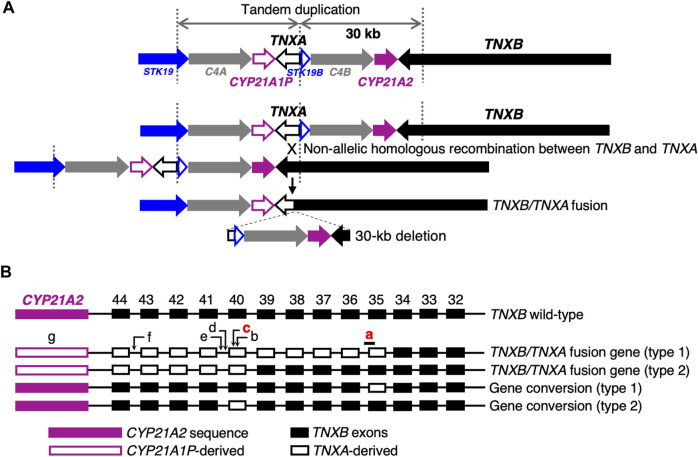
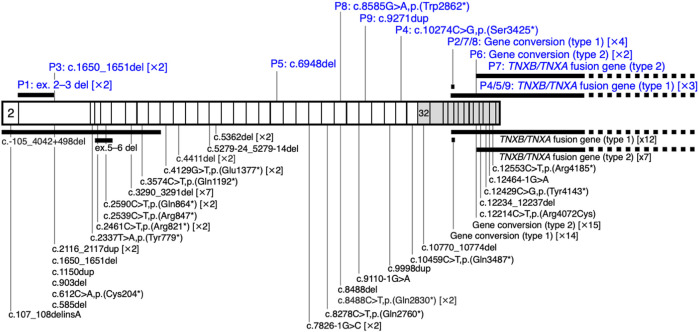
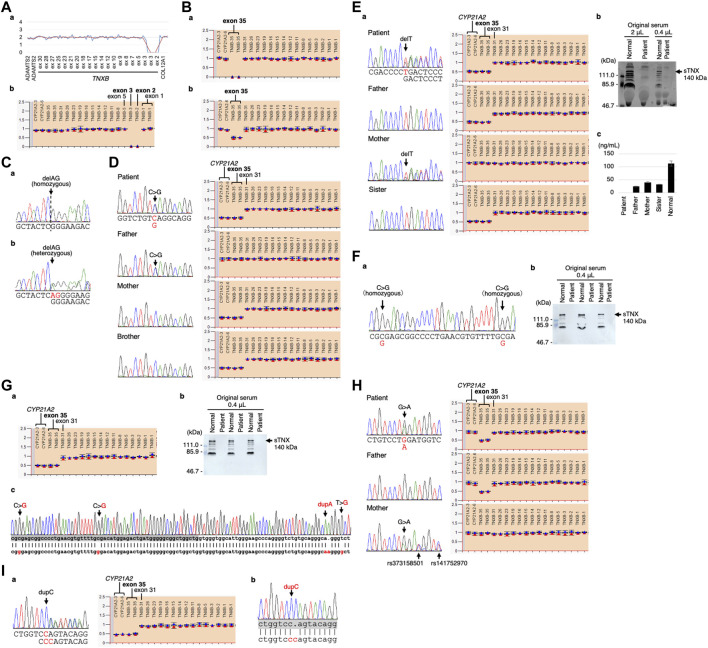
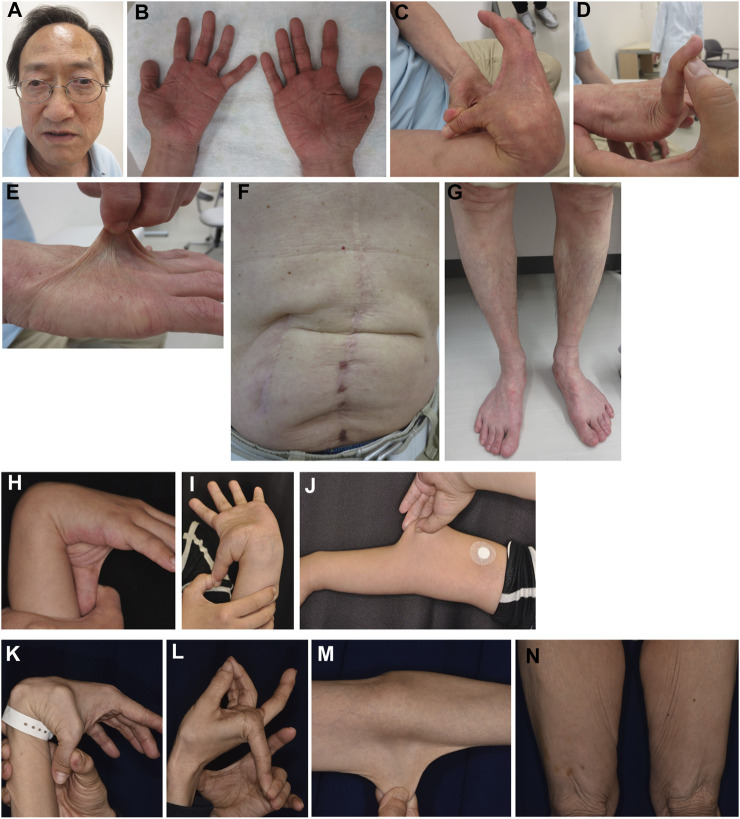
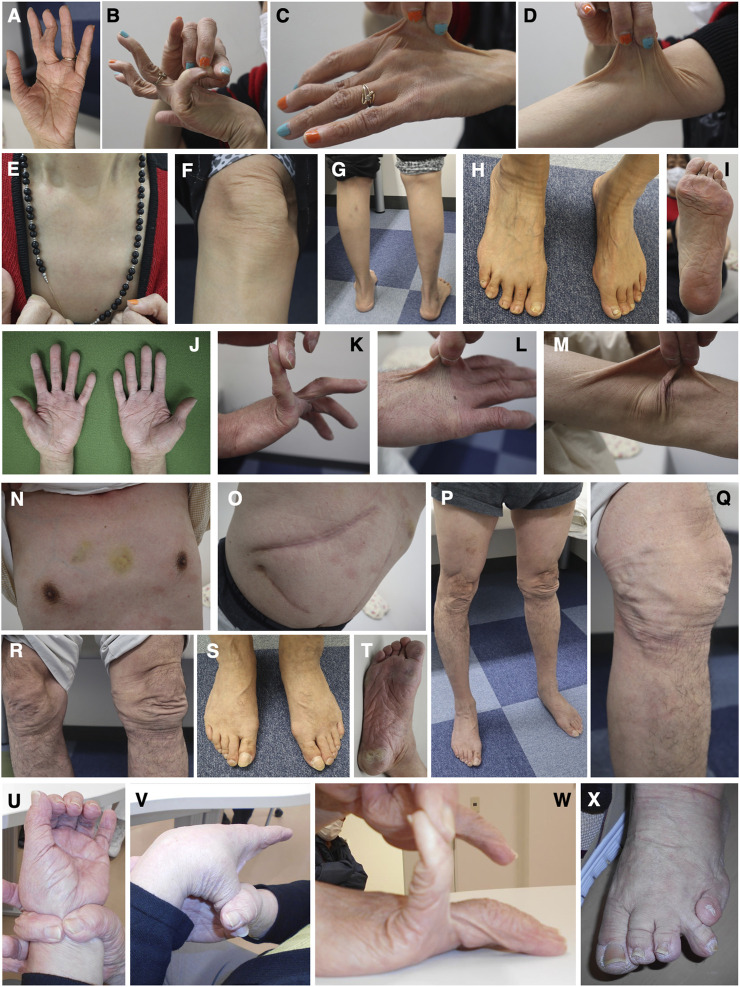
Classical-like Ehlers-Danlos syndrome (clEDS) is an autosomal recessive disorder caused by complete absence of tenascin-X resulting from biallelic variation in TNXB. Thus far, 50 patients from 43 families with biallelic TNXB variants have been identified. Accurate detection of TNXB variants is challenging because of the presence of the pseudogene TNXA, which can undergo non-allelic homologous recombination. Therefore, we designed a genetic screening system that is performed using similar operations to other next-generation sequencing (NGS) panel analyses and can be applied to accurately detect TNXB variants and the recombination of TNXA-derived sequences into TNXB. Using this system, we identified biallelic TNXB variants in nine unrelated clEDS patients. TNXA-derived variations were found in >75% of the current cohort, comparable to previous reports. The current cohort generally exhibited similar clinical features to patients in previous reports, but had a higher frequency of gastrointestinal complications (e.g., perforation, diverticulitis, gastrointestinal bleeding, intestinal obstruction, rectal/anal prolapse, and gallstones). This report is the first to apply an NGS-based screening for TNXB variants and represents the third largest cohort of clEDS, highlighting the importance of increasing awareness of the risk of gastrointestinal complications.
Keywords: Ehlers-Danlos syndrome; TNXB; classical-like; connective tissue disorder; tenascin-X.
Copyright © 2023 Yamaguchi, Yamada, Nagai, Nishikubo, Koitabashi, Minami-Hori, Matsushima, Shibata, Ishiguro, Sanai, Fujikawa, Takiguchi, Matsumoto and Kosho.
Conflict of interest statement
TY, TF, YT, and TK are members of the endowed chair named “Division of Clinical Sequencing, Shinshu University School of Medicine”, which is sponsored by BML, Inc. and Life Technologies Japan Ltd., a subsidiary of Thermo Fisher Scientific Inc. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
TNXB-Related Classical-Like Ehlers-Danlos Syndrome.2022 Sep 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2022 Sep 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 36108117 Free Books & Documents. Review.
-
Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome.Genes (Basel). 2019 Nov 25;10(12):967. doi: 10.3390/genes10120967. Genes (Basel). 2019. PMID: 31775249 Free PMC article.
-
Recurrent pneumothorax in a case of tenascin-X deficient Ehlers-Danlos syndrome: Broadening the phenotypic spectrum.Am J Med Genet A. 2022 May;188(5):1583-1588. doi: 10.1002/ajmg.a.62674. Epub 2022 Feb 6. Am J Med Genet A. 2022. PMID: 35128805 Free PMC article.
-
Broadening the Spectrum of Ehlers Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia.J Clin Endocrinol Metab. 2015 Aug;100(8):E1143-52. doi: 10.1210/jc.2015-2232. Epub 2015 Jun 15. J Clin Endocrinol Metab. 2015. PMID: 26075496 Free PMC article.
-
Congenital Adrenal Hyperplasia and Ehlers-Danlos Syndrome.Front Endocrinol (Lausanne). 2022 Feb 25;13:803226. doi: 10.3389/fendo.2022.803226. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35282436 Free PMC article. Review.
Cited by
-
Clinical diagnosis of the monogenic Ehlers-Danlos syndromes.Med Genet. 2024 Dec 3;36(4):225-234. doi: 10.1515/medgen-2024-2060. eCollection 2024 Dec. Med Genet. 2024. PMID: 39629459 Free PMC article.
-
Genetic diagnosis of the Ehlers-Danlos syndromes.Med Genet. 2024 Dec 3;36(4):235-245. doi: 10.1515/medgen-2024-2061. eCollection 2024 Dec. Med Genet. 2024. PMID: 39629471 Free PMC article.
References
-
- Al-Harbi T. M., Al-Rammah H., Al-Zahrani N., Liu Y., Sleiman P. M. A., Dridi W., et al. (2022). Rare neurological manifestations in a Saudi Arabian patient with Ehlers–Danlos syndrome and a novel homozygous variant in the TNXB gene. Am. J. Med. Genet. A 188 (2), 618–623. 10.1002/ajmg.a.62539 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous