Integrated BATF transcriptional network regulates suppressive intratumoral regulatory T cells
- PMID: 37713508
- PMCID: PMC11045170
- DOI: 10.1126/sciimmunol.adf6717
Integrated BATF transcriptional network regulates suppressive intratumoral regulatory T cells
Abstract
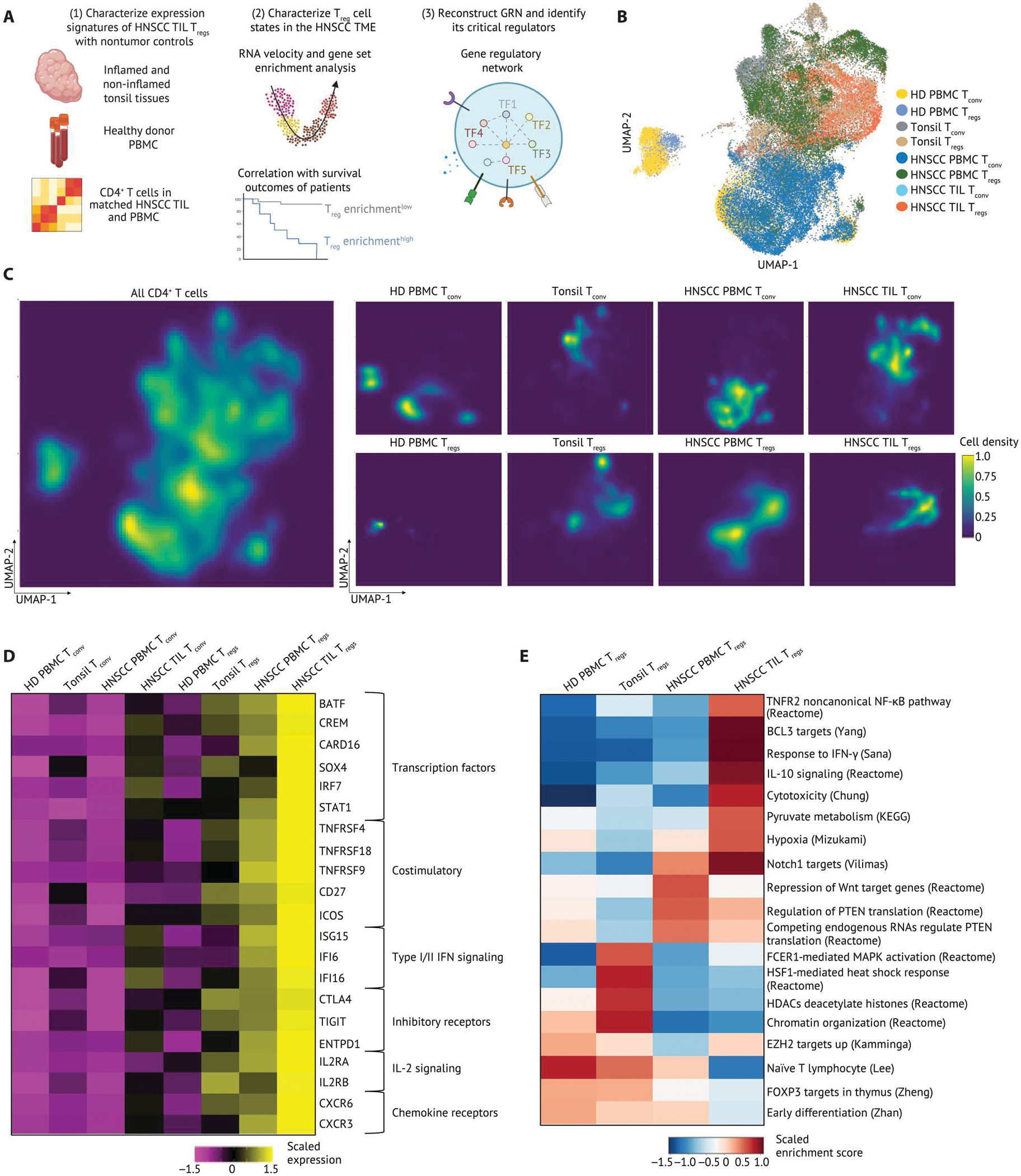
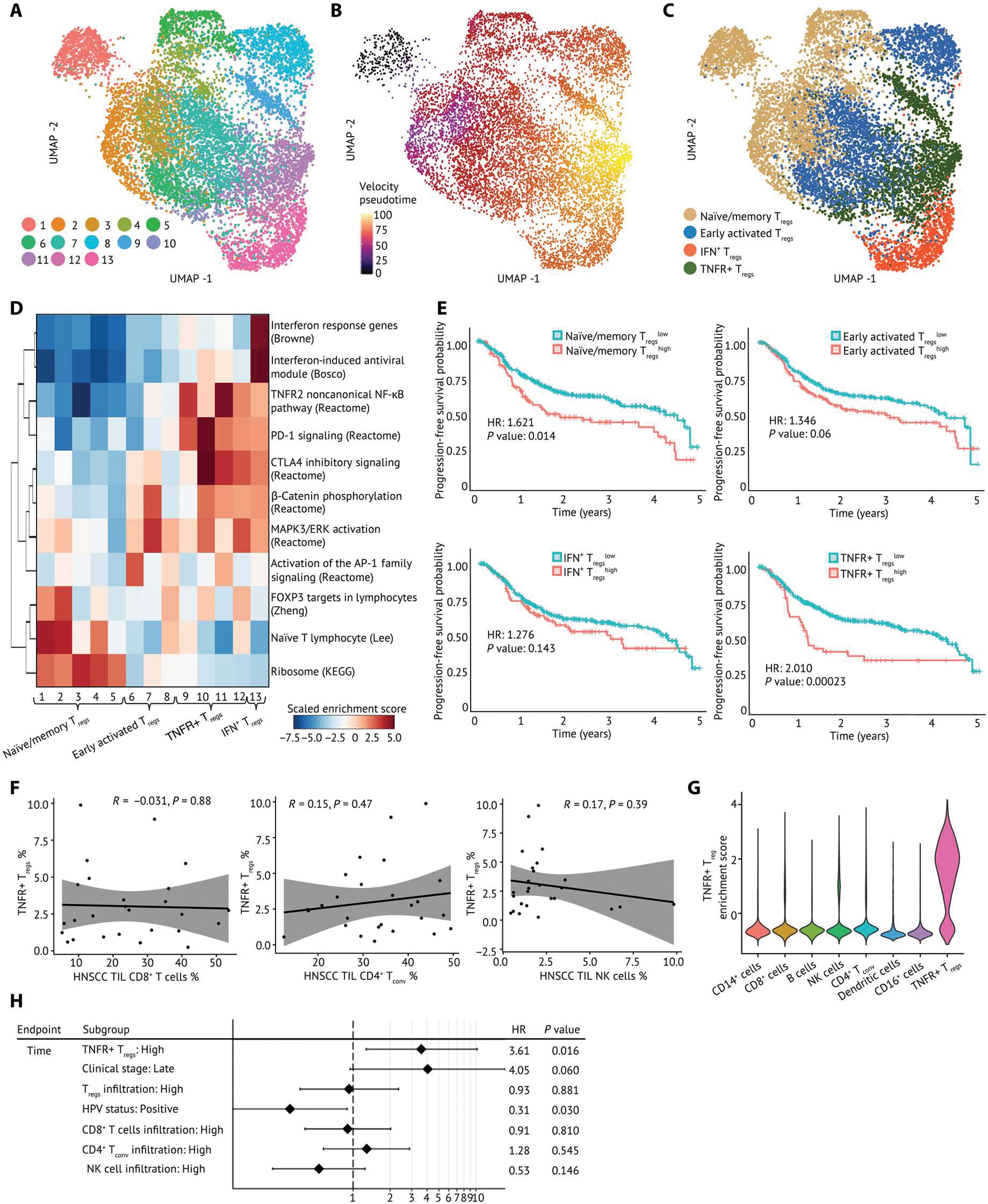
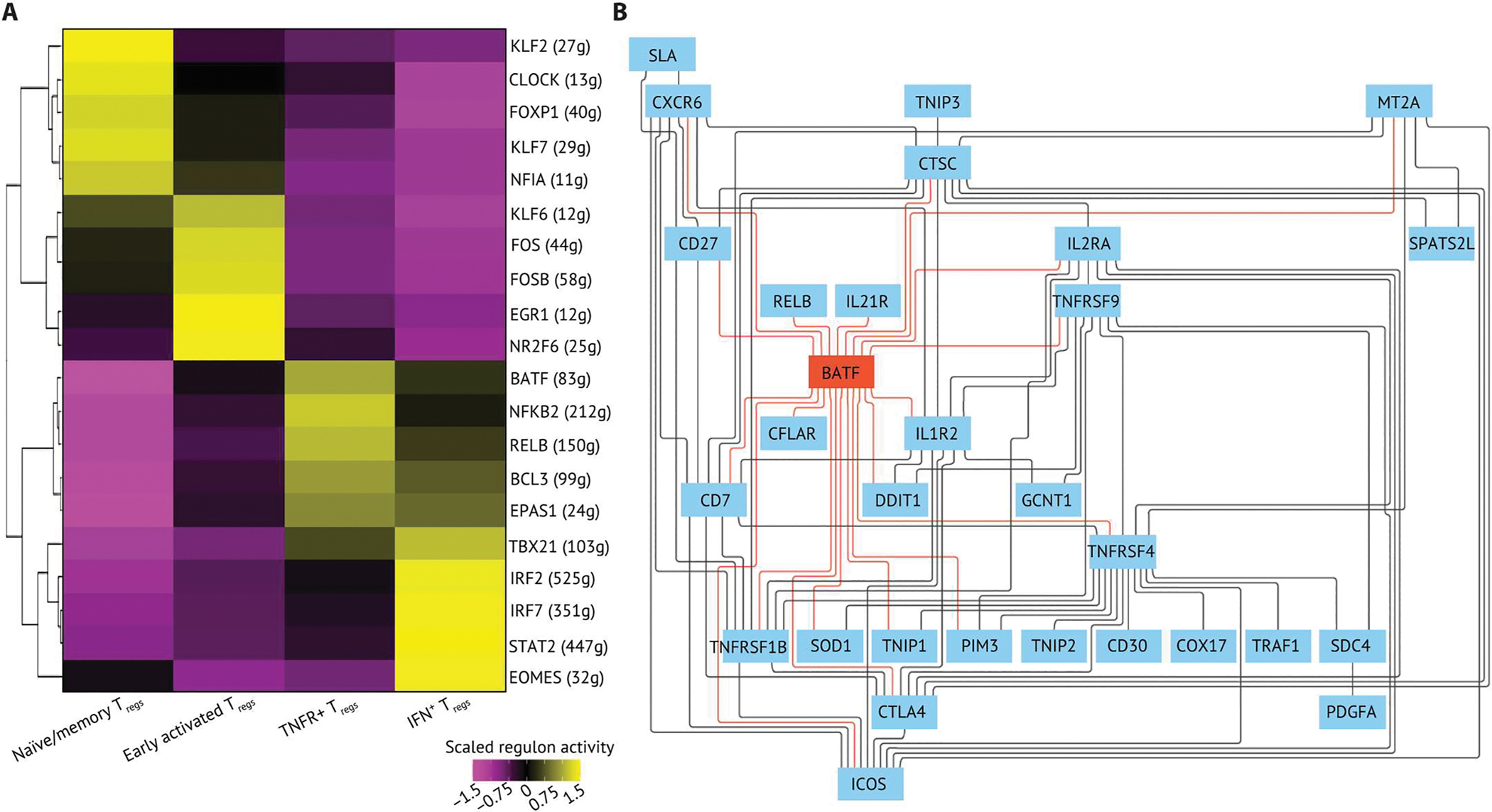
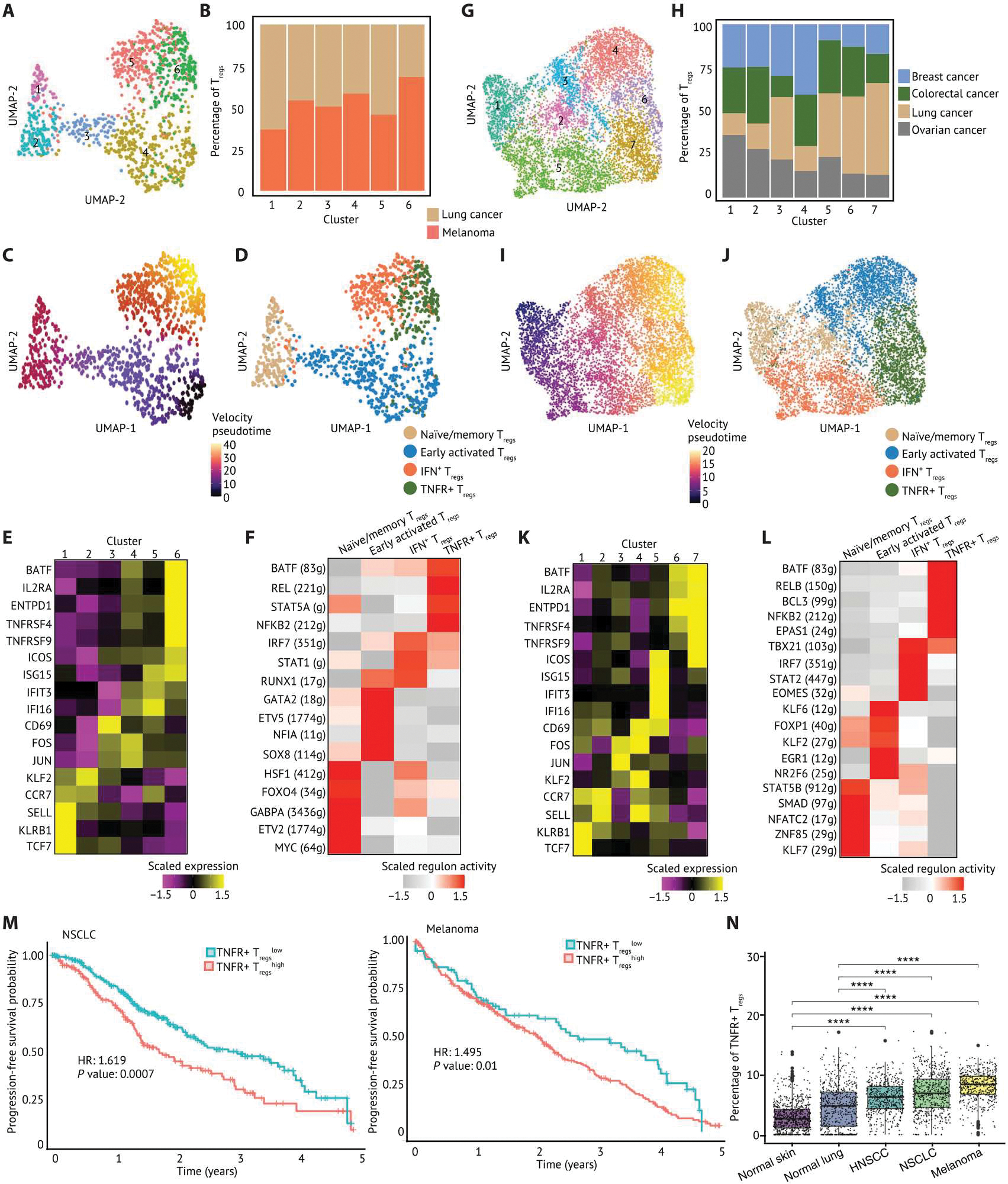
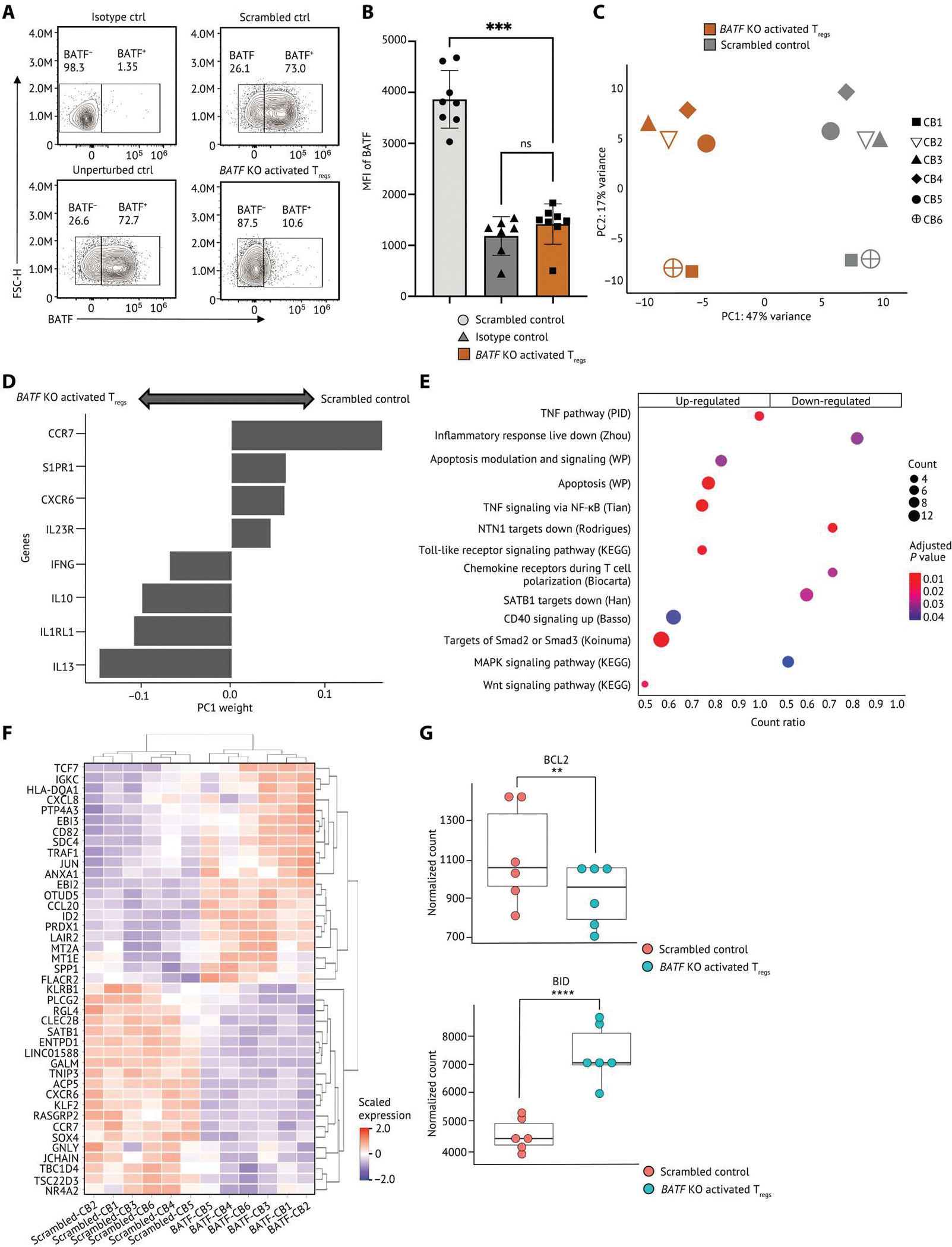
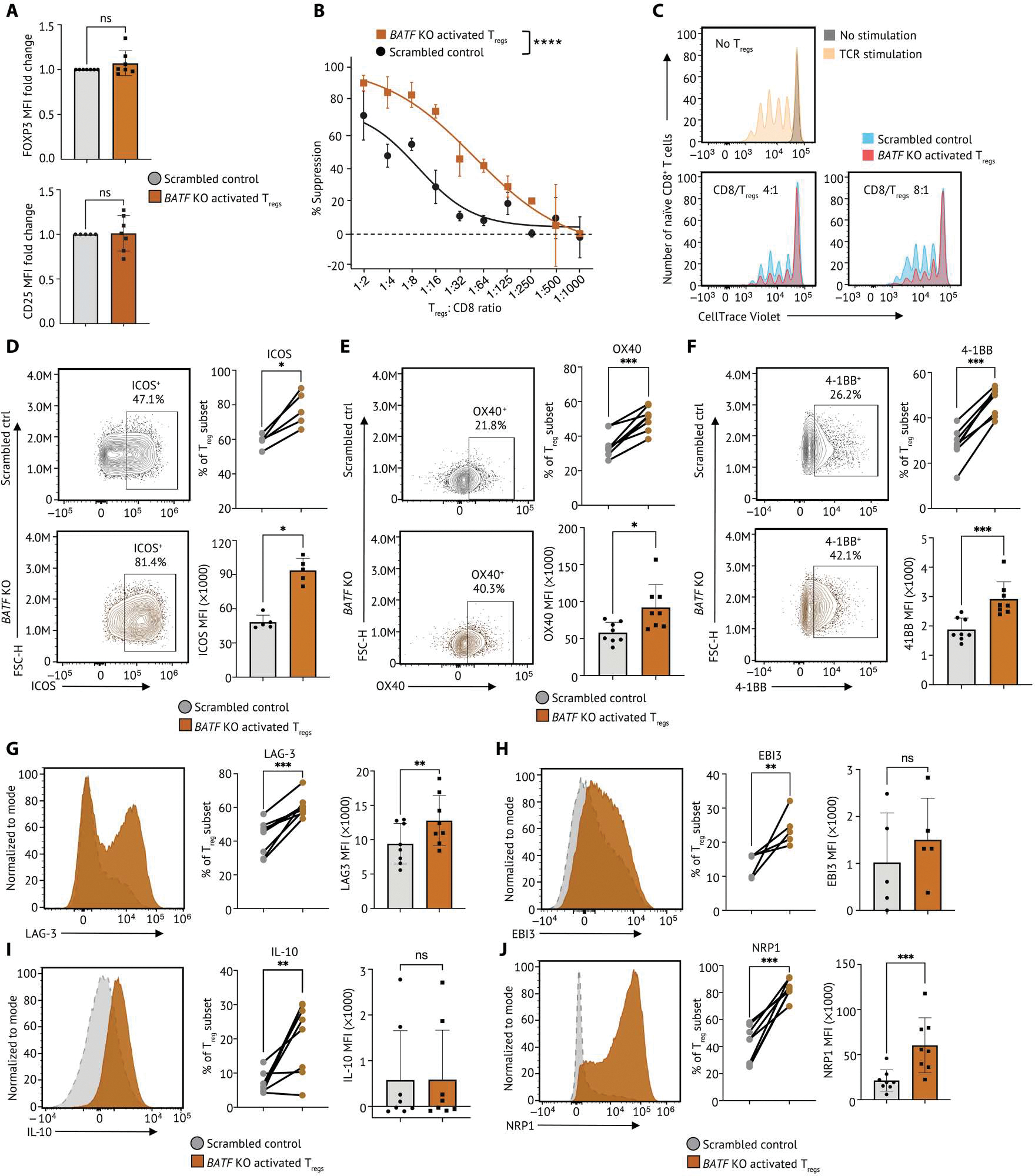
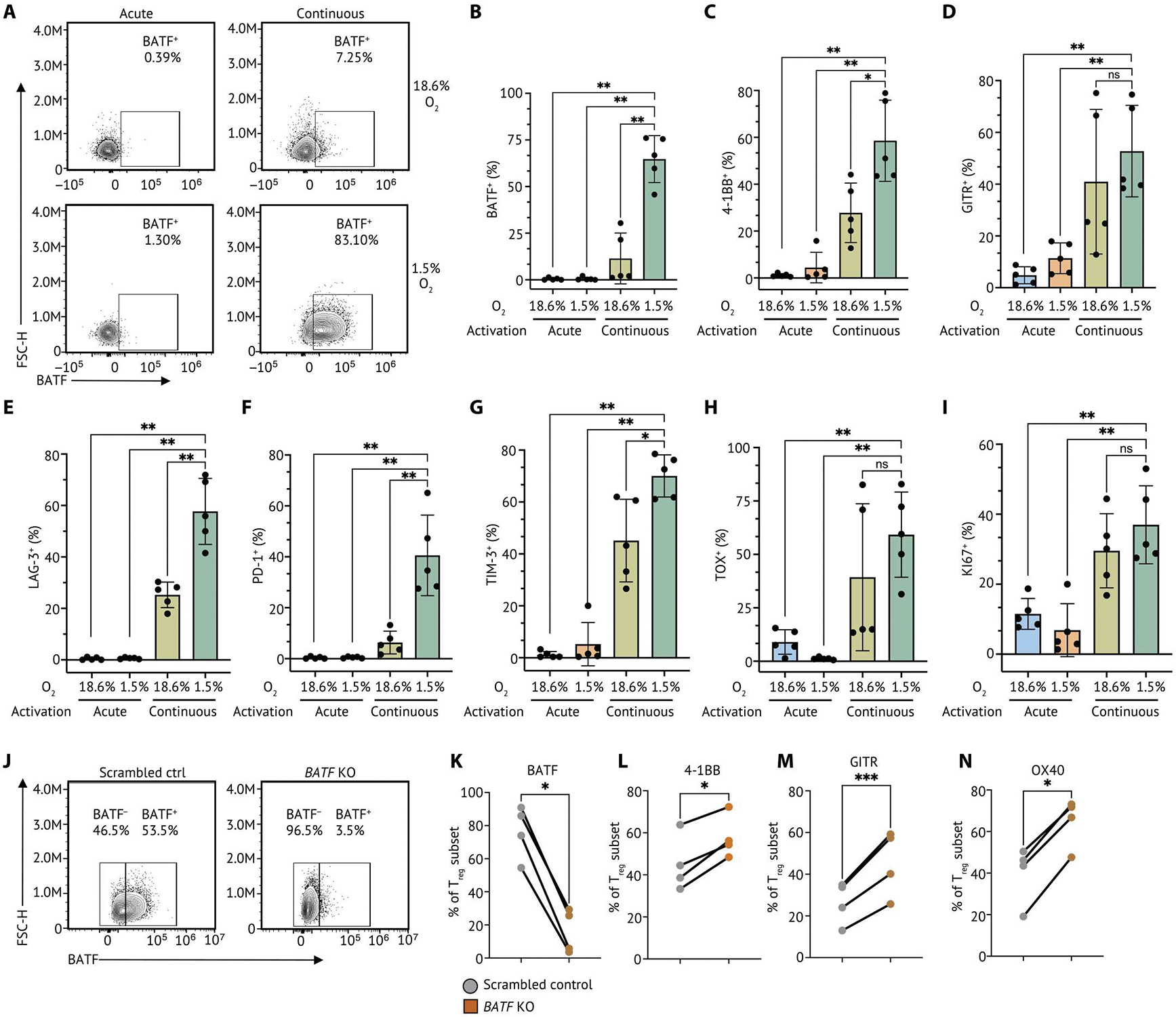
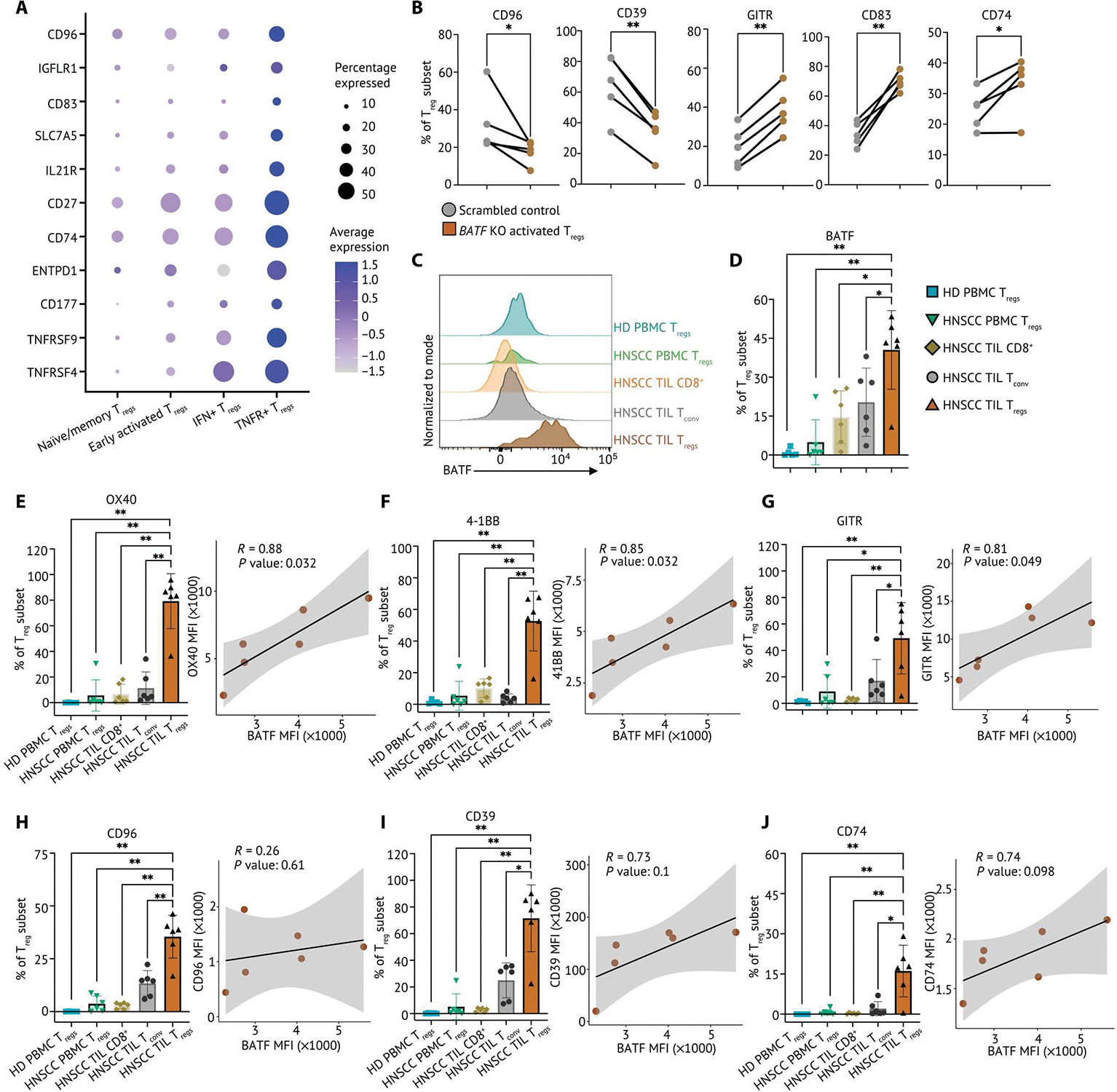
Human regulatory T cells (Tregs) are crucial regulators of tissue repair, autoimmune diseases, and cancer. However, it is challenging to inhibit the suppressive function of Tregs for cancer therapy without affecting immune homeostasis. Identifying pathways that may distinguish tumor-restricted Tregs is important, yet the transcriptional programs that control intratumoral Treg gene expression, and that are distinct from Tregs in healthy tissues, remain largely unknown. We profiled single-cell transcriptomes of CD4+ T cells in tumors and peripheral blood from patients with head and neck squamous cell carcinomas (HNSCC) and those in nontumor tonsil tissues and peripheral blood from healthy donors. We identified a subpopulation of activated Tregs expressing multiple tumor necrosis factor receptor (TNFR) genes (TNFR+ Tregs) that is highly enriched in the tumor microenvironment (TME) compared with nontumor tissue and the periphery. TNFR+ Tregs are associated with worse prognosis in HNSCC and across multiple solid tumor types. Mechanistically, the transcription factor BATF is a central component of a gene regulatory network that governs key aspects of TNFR+ Tregs. CRISPR-Cas9-mediated BATF knockout in human activated Tregs in conjunction with bulk RNA sequencing, immunophenotyping, and in vitro functional assays corroborated the central role of BATF in limiting excessive activation and promoting the survival of human activated Tregs. Last, we identified a suite of surface molecules reflective of the BATF-driven transcriptional network on intratumoral Tregs in patients with HNSCC. These findings uncover a primary transcriptional regulator of highly suppressive intratumoral Tregs, highlighting potential opportunities for therapeutic intervention in cancer without affecting immune homeostasis.
Conflict of interest statement
Figures
References
-
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21 (2001). - PubMed
-
- Liu C, Workman CJ, Vignali DAA, Targeting regulatory T cells in tumors. FEBS J. 283, 2731–2748 (2016). - PubMed
-
- Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z, Nielsen TO, Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 16, 432 (2014). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials