

TGF-β signaling in health and disease
- PMID: 37714133
- PMCID: PMC10772989
- DOI: 10.1016/j.cell.2023.07.036
TGF-β signaling in health and disease
Abstract
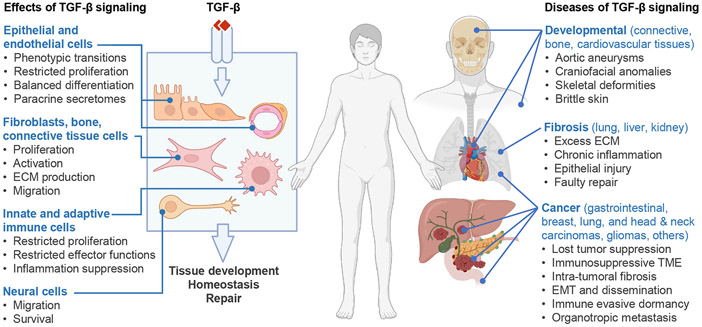
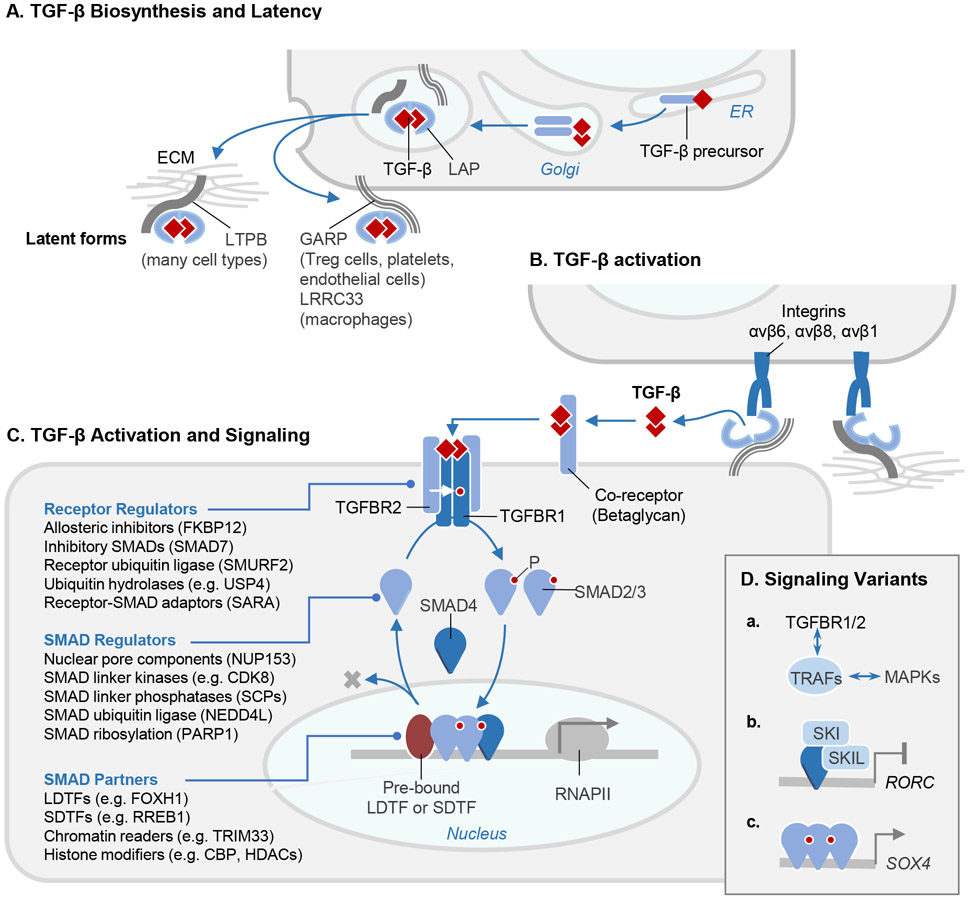
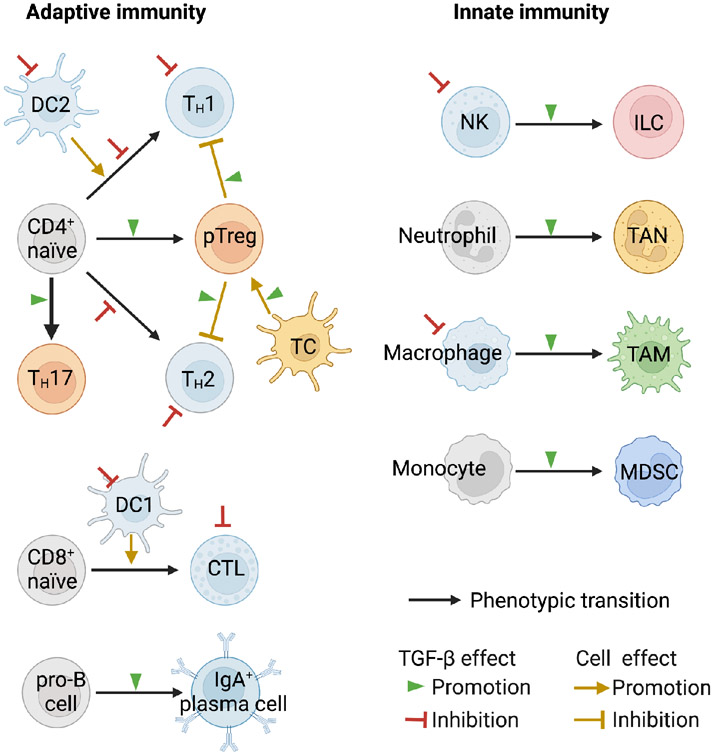
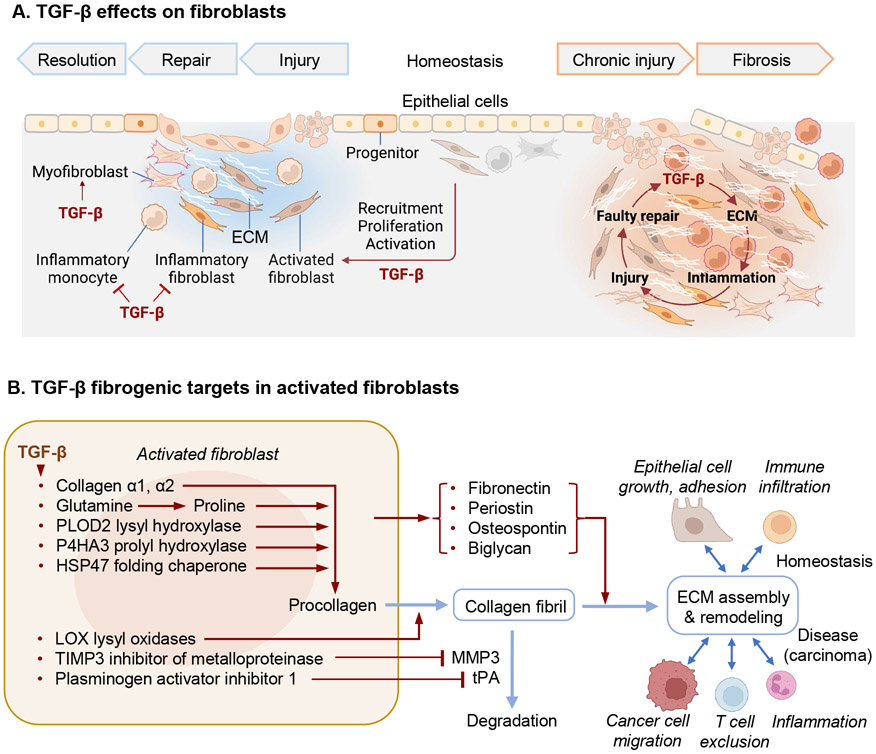
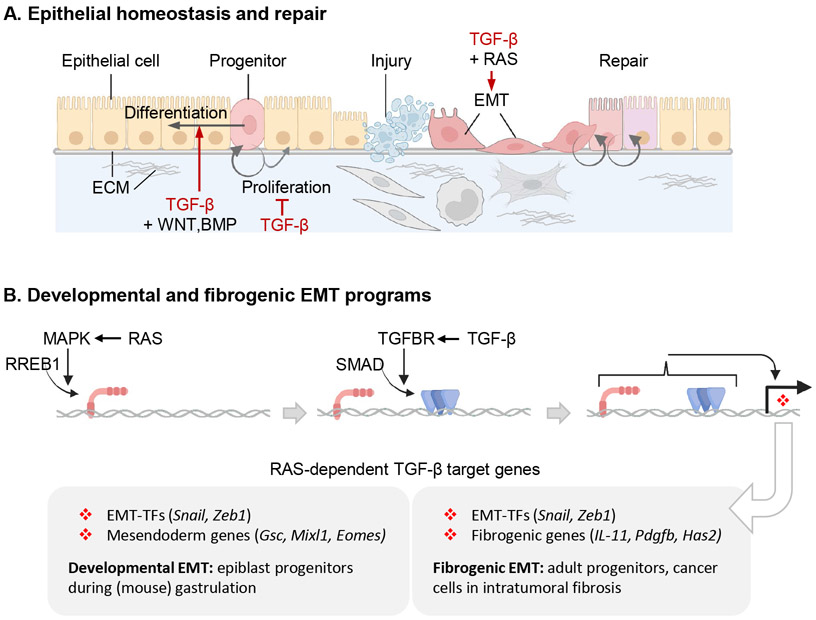
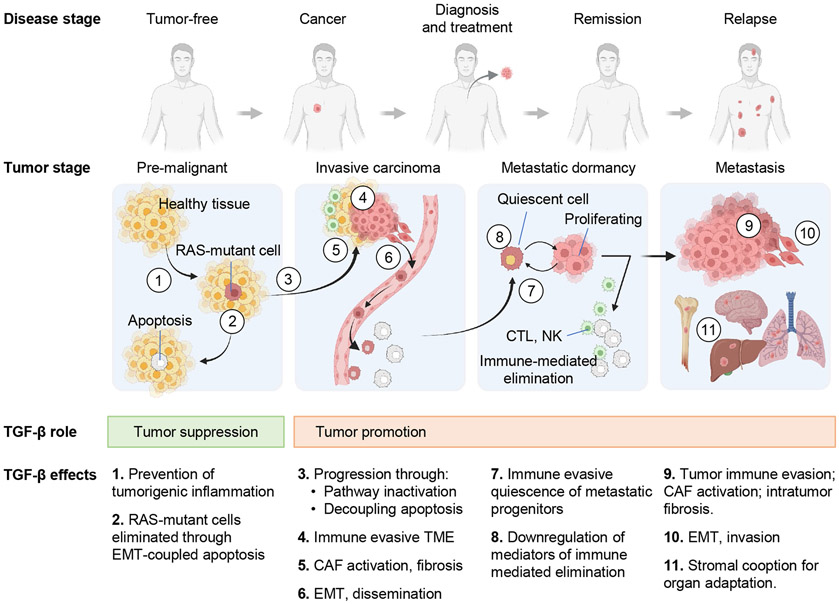
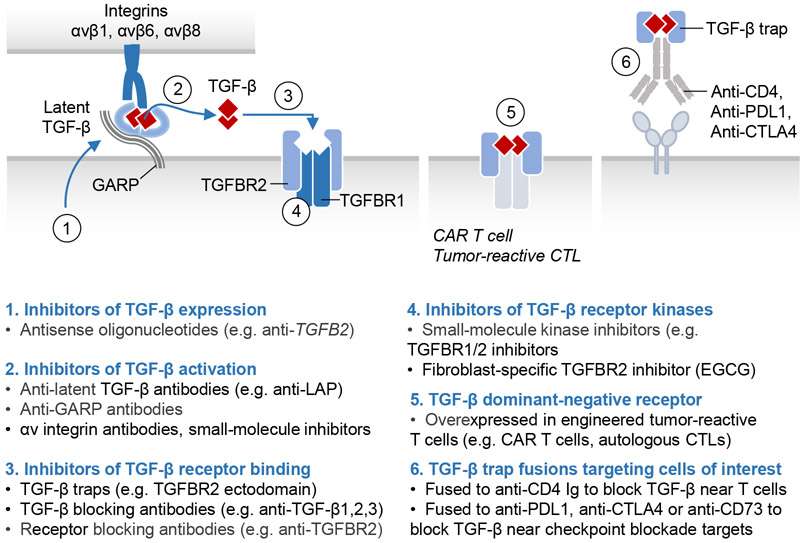
The TGF-β regulatory system plays crucial roles in the preservation of organismal integrity. TGF-β signaling controls metazoan embryo development, tissue homeostasis, and injury repair through coordinated effects on cell proliferation, phenotypic plasticity, migration, metabolic adaptation, and immune surveillance of multiple cell types in shared ecosystems. Defects of TGF-β signaling, particularly in epithelial cells, tissue fibroblasts, and immune cells, disrupt immune tolerance, promote inflammation, underlie the pathogenesis of fibrosis and cancer, and contribute to the resistance of these diseases to treatment. Here, we review how TGF-β coordinates multicellular response programs in health and disease and how this knowledge can be leveraged to develop treatments for diseases of the TGF-β system.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.M. holds company stock of Scholar Rock, Inc. D.S. is a founder of Pliant Therapeutics, a member of the Genentech Scientific Review Board, a member of the Amgen Immunology Scientific Advisory Board, and a member of the Scientific Review Board for Lila Biologics.
Figures
References
-
- Roberts AB, and Sporn MB (1988). Transforming growth factor beta. Adv Cancer Res 51, 107–145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
