

First fully-automated AI/ML virtual screening cascade implemented at a drug discovery centre in Africa
- PMID: 37714843
- PMCID: PMC10504240
- DOI: 10.1038/s41467-023-41512-2
First fully-automated AI/ML virtual screening cascade implemented at a drug discovery centre in Africa
Abstract
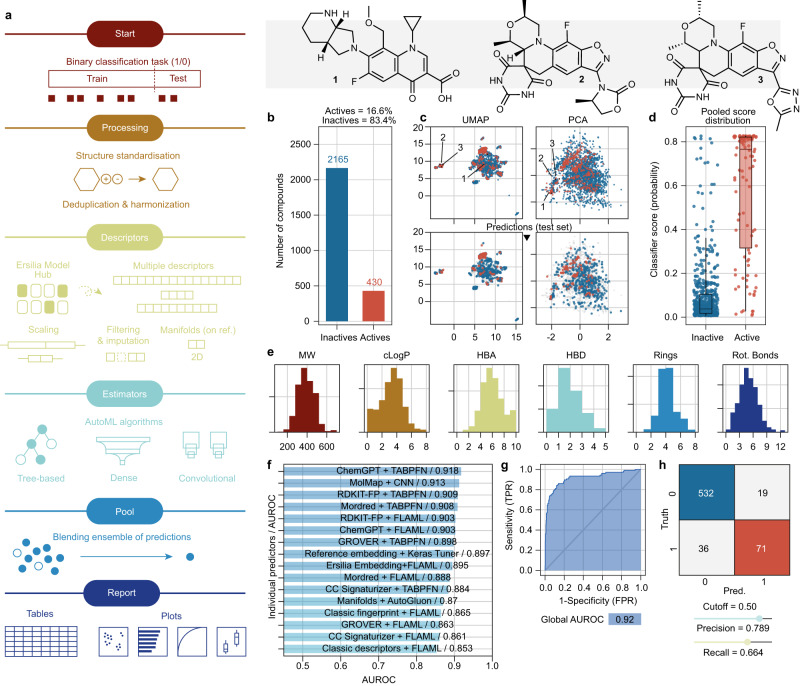
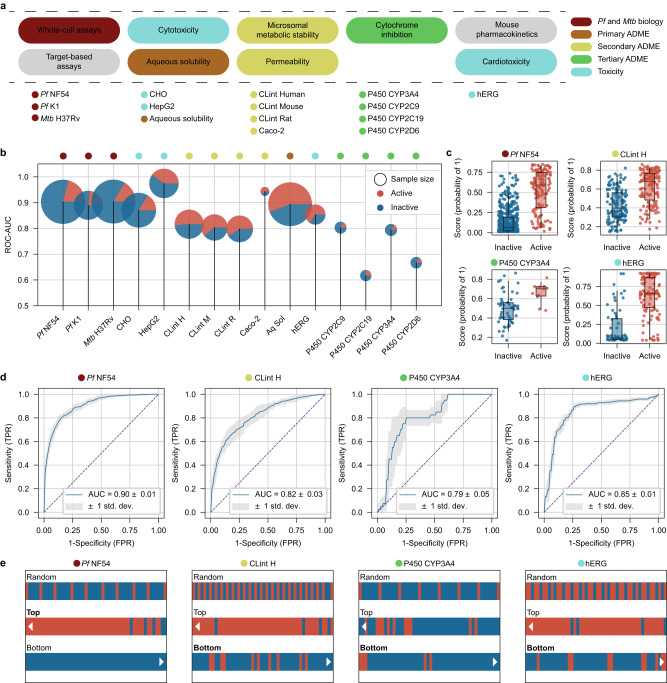
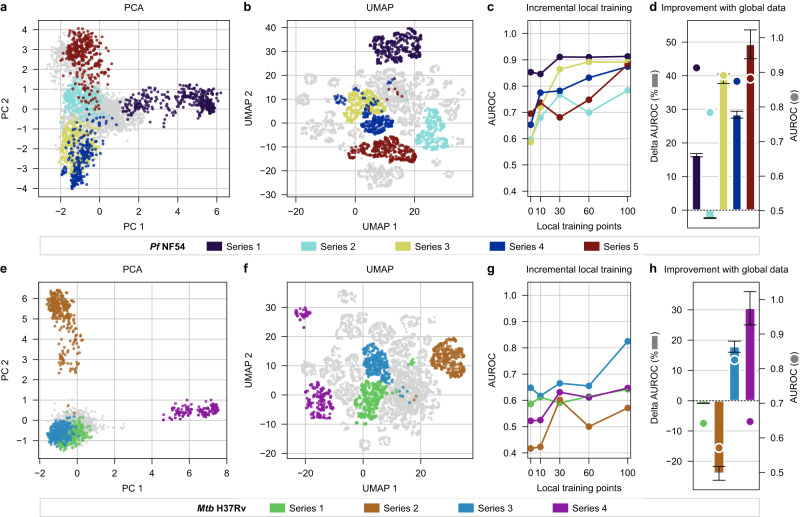
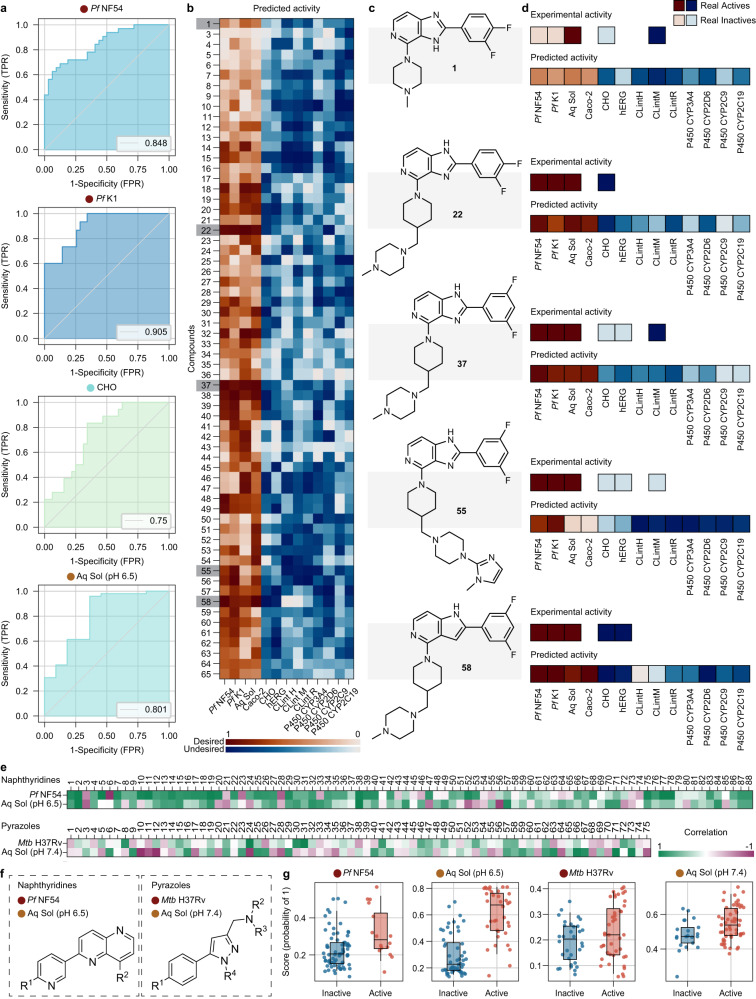
Streamlined data-driven drug discovery remains challenging, especially in resource-limited settings. We present ZairaChem, an artificial intelligence (AI)- and machine learning (ML)-based tool for quantitative structure-activity/property relationship (QSAR/QSPR) modelling. ZairaChem is fully automated, requires low computational resources and works across a broad spectrum of datasets. We describe an end-to-end implementation at the H3D Centre, the leading integrated drug discovery unit in Africa, at which no prior AI/ML capabilities were available. By leveraging in-house data collected over a decade, we have developed a virtual screening cascade for malaria and tuberculosis drug discovery comprising 15 models for key decision-making assays ranging from whole-cell phenotypic screening and cytotoxicity to aqueous solubility, permeability, microsomal metabolic stability, cytochrome inhibition, and cardiotoxicity. We show how computational profiling of compounds, prior to synthesis and testing, can inform progression of frontrunner compounds at H3D. This project is a first-of-its-kind deployment at scale of AI/ML tools in a research centre operating in a low-resource setting.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kirkpatrick, P. Artificial intelligence makes a splash in small-molecule drug discovery. Biopharma Dealmakers10.1038/d43747-022-00104-7 (2022).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
