

Efficacy of a bivalent (D614 + B.1.351) SARS-CoV-2 recombinant protein vaccine with AS03 adjuvant in adults: a phase 3, parallel, randomised, modified double-blind, placebo-controlled trial
- PMID: 37716365
- PMCID: PMC10872639
- DOI: 10.1016/S2213-2600(23)00263-1
Efficacy of a bivalent (D614 + B.1.351) SARS-CoV-2 recombinant protein vaccine with AS03 adjuvant in adults: a phase 3, parallel, randomised, modified double-blind, placebo-controlled trial
Abstract
Background: COVID-19 vaccines with alternative strain compositions are needed to provide broad protection against newly emergent SARS-CoV-2 variants of concern. This study aimed to describe the clinical efficacy and safety of a bivalent SARS-CoV-2 recombinant protein vaccine as a two-injection primary series during a period of circulation of the omicron (B.1.1.529) variant.
Methods: We conducted a phase 3, parallel, randomised, modified double-blind, placebo-controlled trial in adults aged 18 years or older at 54 clinical research centres in eight countries (Colombia, Ghana, India, Kenya, Mexico, Nepal, Uganda, and Ukraine). Participants were recruited from the community and randomly assigned (1:1) by use of an interactive response technology system to receive two intramuscular 0·5 mL injections, 21 days apart, of the bivalent vaccine (5 μg of ancestral [D614] and 5 μg of beta [B.1.351] variant spike protein, with AS03 adjuvant) or placebo (0·9% normal saline). All participants, outcome assessors, and laboratory staff performing assays were masked to group assignments; those involved in the preparation and administration of the vaccines were unmasked. Participants were stratified by age (18-59 years and ≥60 years) and baseline SARS-CoV-2 rapid serodiagnostic test positivity. Symptomatic COVID-19 was defined as laboratory-confirmed (via nucleic acid amplification test or PCR test) COVID-19 with COVID-19-like illness symptoms. The primary efficacy endpoint was the clinical efficacy of the bivalent vaccine for prevention of symptomatic COVID-19 at least 14 days after the second injection (dose 2). Safety was assessed in all participants receiving at least one injection of the study vaccine or placebo. This trial is registered with ClinicalTrials.gov (NCT04904549) and is closed to recruitment.
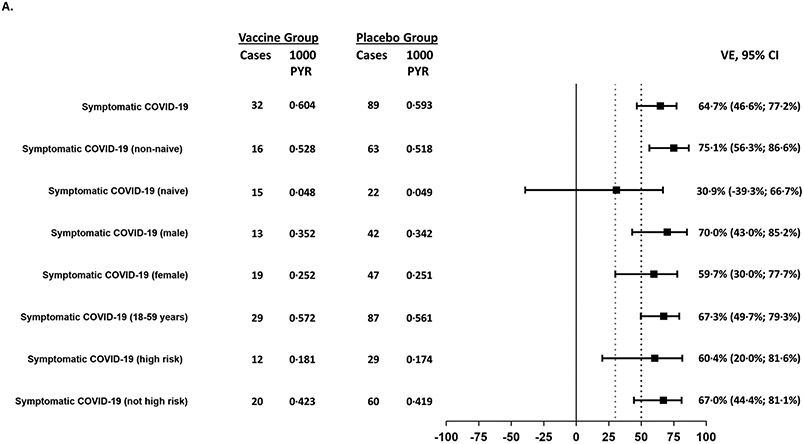
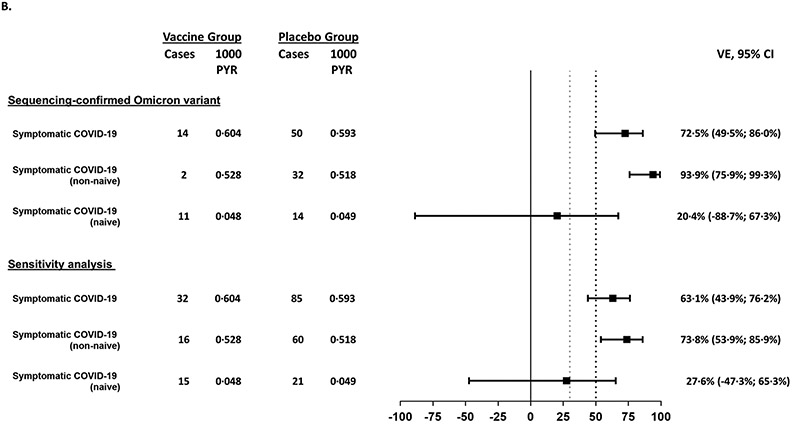
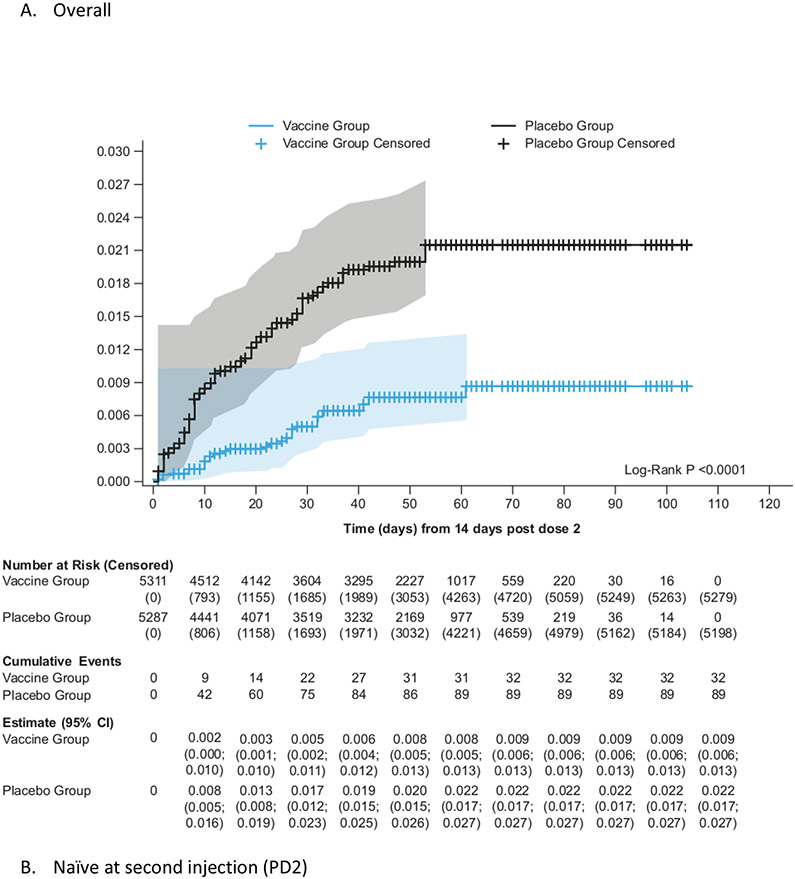
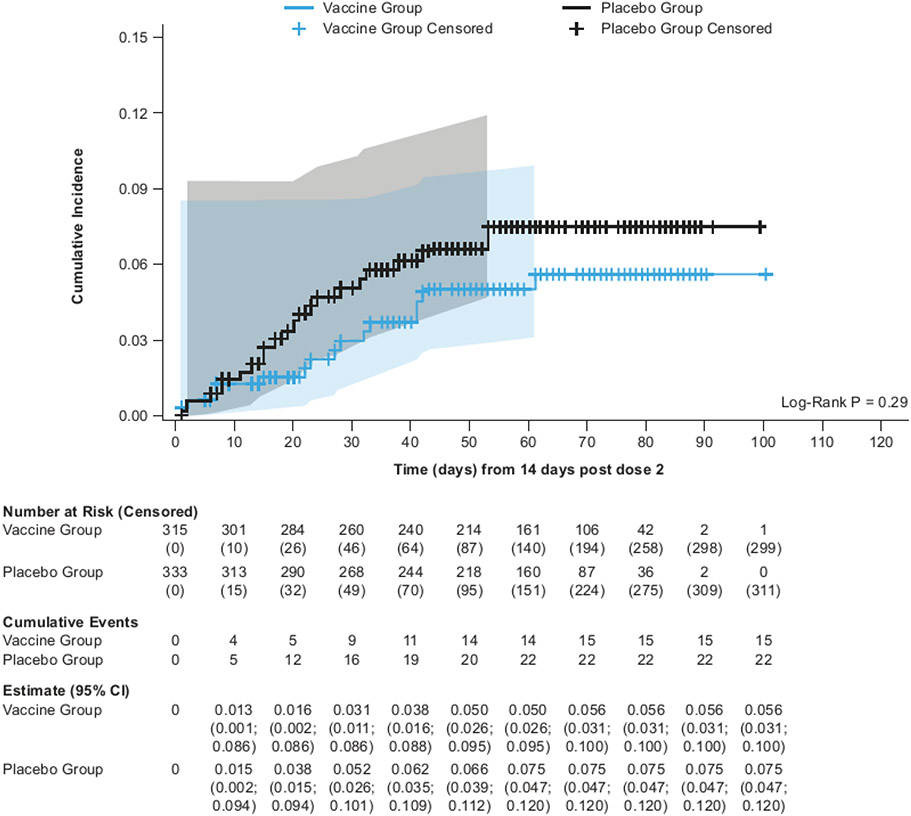
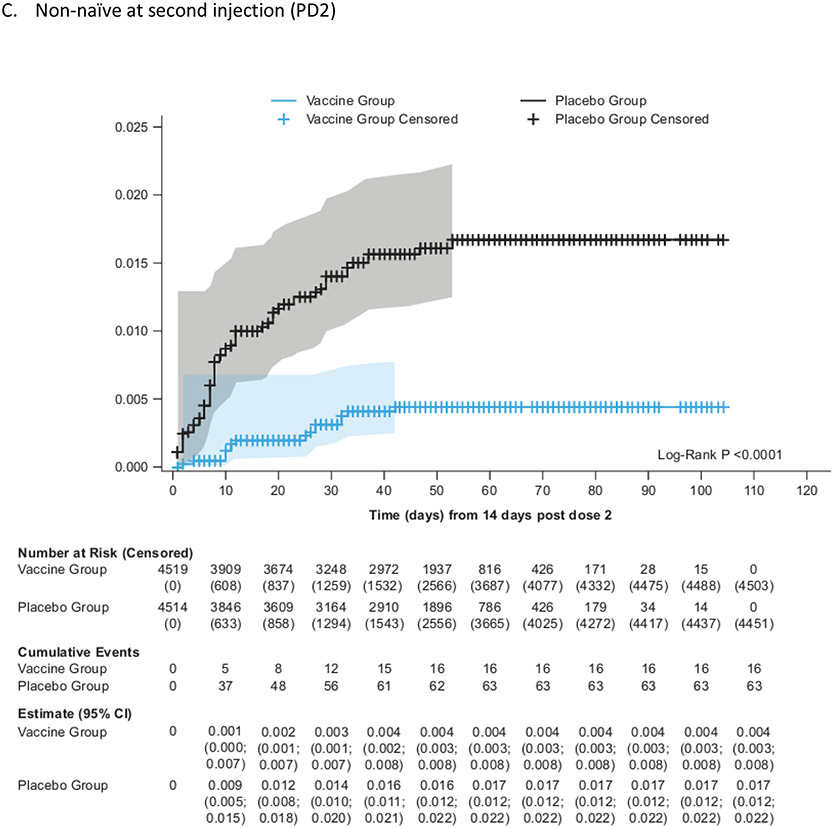
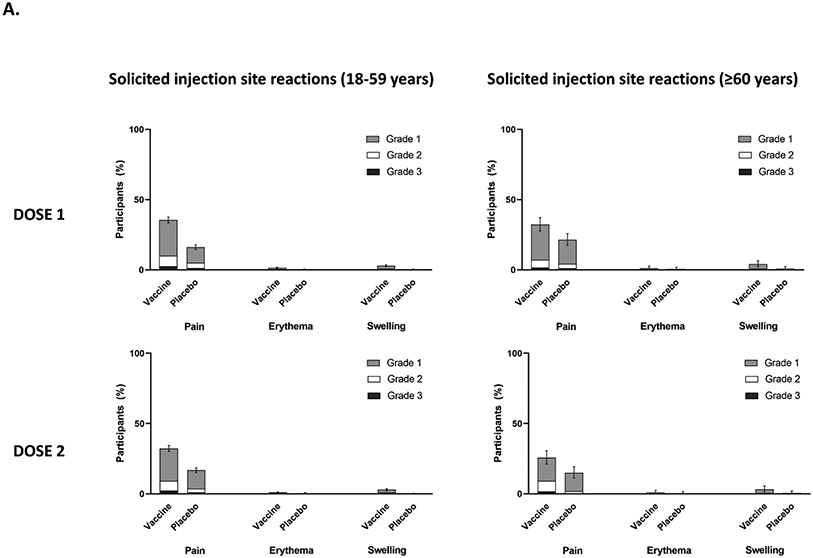
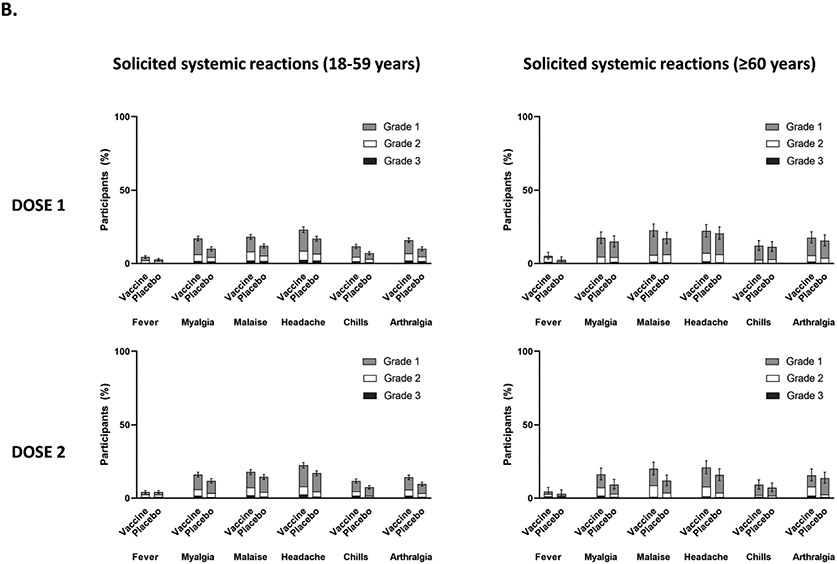
Findings: Between Oct 19, 2021, and Feb 15, 2022, 13 002 participants were enrolled and randomly assigned to receive the first dose of the study vaccine (n=6512) or placebo (n=6490). 12 924 participants (6472 in the vaccine group and 6452 in the placebo group) received at least one study injection, of whom 7542 (58·4%) were male and 9693 (75·0%) were SARS-CoV-2 non-naive. Of these 12 924 participants, 11 543 (89·3%) received both study injections (5788 in the vaccine group and 5755 in the placebo group). The efficacy-evaluable population after dose 2 comprised 11 416 participants (5736 in the vaccine group and 5680 in the placebo group). The median duration of follow-up was 85 days (IQR 50-95) after dose 1 and 58 days (29-70) after dose 2. 121 symptomatic COVID-19 cases were reported at least 14 days after dose 2 (32 in the vaccine group and 89 in the placebo group), with an overall vaccine efficacy of 64·7% (95% CI 46·6 to 77·2). Vaccine efficacy against symptomatic COVID-19 was 75·1% (95% CI 56·3 to 86·6) in SARS-CoV-2 non-naive participants and 30·9% (-39·3 to 66·7) in SARS-CoV-2-naive participants. Viral genome sequencing identified the infecting strain in 68 (56·2%) of 121 cases (omicron [BA.1 and BA.2] in 63; delta in four; and both omicron and delta in one). Immediate unsolicited adverse events were reported by four (<0·1%) participants in the vaccine group and seven (0·1%) participants in the placebo group. Immediate unsolicited adverse reactions within 30 min after any injection were reported by four (<0·1%) participants in the vaccine group and six (<0·1%) participants in the placebo group. In the reactogenicity subset with available data, solicited reactions (solicited injection-site reactions and solicited systemic reactions) within 7 days after any injection occurred in 1398 (57·8%) of 2420 vaccine recipients and 983 (40·9%) of 2403 placebo recipients. Grade 3 solicited reactions were reported by 196 (8·1%; 95% CI 7·0 to 9·3) of 2420 vaccine recipients and 118 (4·9%; 4·1 to 5·9) of 2403 placebo recipients within 7 days after any injection, with comparable frequencies after dose 1 and dose 2 in the vaccine group. At least one serious adverse event occurred in 30 (0·5%) participants in the vaccine group and 26 (0·4%) in the placebo group. The proportion of adverse events of special interest and deaths was less than 0·1% in both study groups. No adverse event of special interest, serious adverse event, or death was deemed to be treatment related. There were no reported cases of thrombosis with thrombocytopenia syndrome, myocarditis, pericarditis, Bell's Palsy, or Guillain-Barré syndrome, or other immune-mediated diseases.
Interpretation: The bivalent variant vaccine conferred heterologous protection against symptomatic SARS-CoV-2 infection in the epidemiological context of the circulating contemporary omicron variant. These findings suggest that vaccines developed with an antigen from a non-predominant strain could confer cross-protection against newly emergent SARS-CoV-2 variants, although further investigation is warranted.
Funding: Sanofi, US Biomedical Advanced Research and Development Authority, and the US National Institute of Allergy and Infectious Diseases.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests GHD, MIB, BF, M-HG, MC, JA, CAD, RMC, SG, SSr, and SSa are Sanofi employees. MIB, BF, M-HG, JA, CAD, RMC, and SSr hold stock or stock options in Sanofi. SG, RMC, and SSr hold patents pending on a COVID-19 vaccine. The Center for Vaccine Development and Global Health (CVD) receives grants from Pfizer to conduct clinical trials of COVID-19 vaccines. KMN receives no salary support for this grant. KMN receives grants from the US National Institutes of Health (NIH) to participate in overall organisation of COVID-19 vaccine trials and for participation in vaccine trials. RMC has received institutional funding from the Biomedical Advanced Research and Development Authority (BARDA) for the present study; has received support for attending meetings or travel, from Sanofi; and holds patents planned, issued, or pending from Sanofi. M-HG has received payment or honoraria for lectures, presentations, speakers' bureaus, manuscript writing, or educational events from Sanofi. NR has received institutional funding from the NIH; and institutional grants or contracts from Merck, Sanofi, Quidel, Pfizer, and Lilly. SRW has received institutional funding from Sanofi and the National Institute of Allergy and Infectious Diseases (NIAID) and NIH; and institutional grants or contracts from Janssen Vaccines/Johnson & Johnson, Moderna Tx, Vir Biotechnology, and Worcester HIV Vaccine; has participated on data safety monitoring or advisory boards for Janssen Vaccines/Johnson & Johnson; and his spouse holds stocks and stock options in Regeneron Pharmaceuticals. NG has received institutional funding from Sanofi, GSK, and the NIAID and NIH; and is in receipt of grants or contracts from the NIH, NIAID, and the Division of AIDS (DAIDS). MA and TT are employees of NIAID, which funded aspects of the current study. LS, MAC, and MK are employees of GSK and own shares in the GSK group of companies. MJ and JJK have received institutional support from Sanofi and NIAID and NIH with respect to this study. MLR has received institutional support or contracts for the present manuscript from Walter Reed Army Institute of Research, Ingenuity Pathway Analysis, and the US Medical Research and Development Command. SSa was a Sanofi employee at the time of study conduct; and holds patents planned, issued, or pending on COVID-19 vaccines. LC has received grant funding from the NIAID/NIH. AC, KPA, ASB, TB, DD, MK-J, HK, RM, NLM, HR, SMVM, FS, JT, and TAW, declare no competing interests.
Figures
Comment in
-
Broad protection from SARS-CoV-2 variants without antigen matching.Lancet Respir Med. 2023 Nov;11(11):947-948. doi: 10.1016/S2213-2600(23)00290-4. Epub 2023 Sep 13. Lancet Respir Med. 2023. PMID: 37716366 No abstract available.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
