

Performance of tumour microenvironment deconvolution methods in breast cancer using single-cell simulated bulk mixtures
- PMID: 37717006
- PMCID: PMC10505141
- DOI: 10.1038/s41467-023-41385-5
Performance of tumour microenvironment deconvolution methods in breast cancer using single-cell simulated bulk mixtures
Abstract
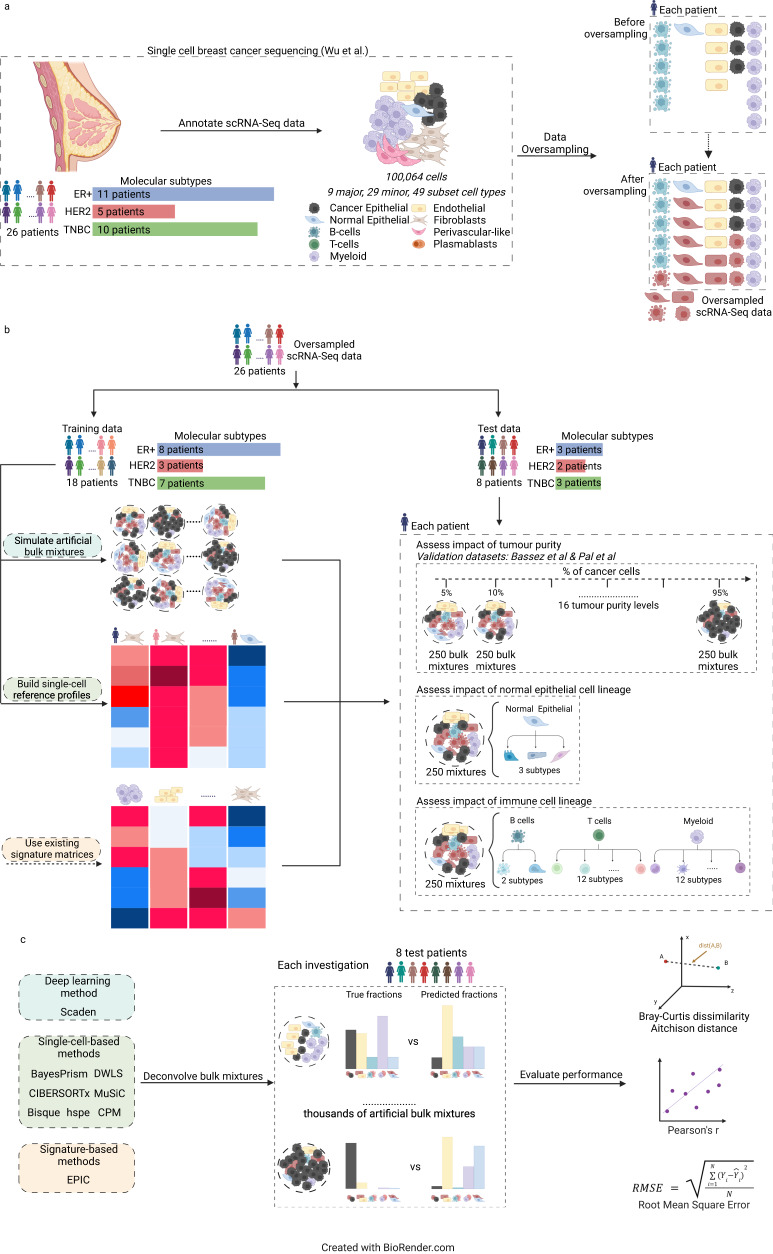
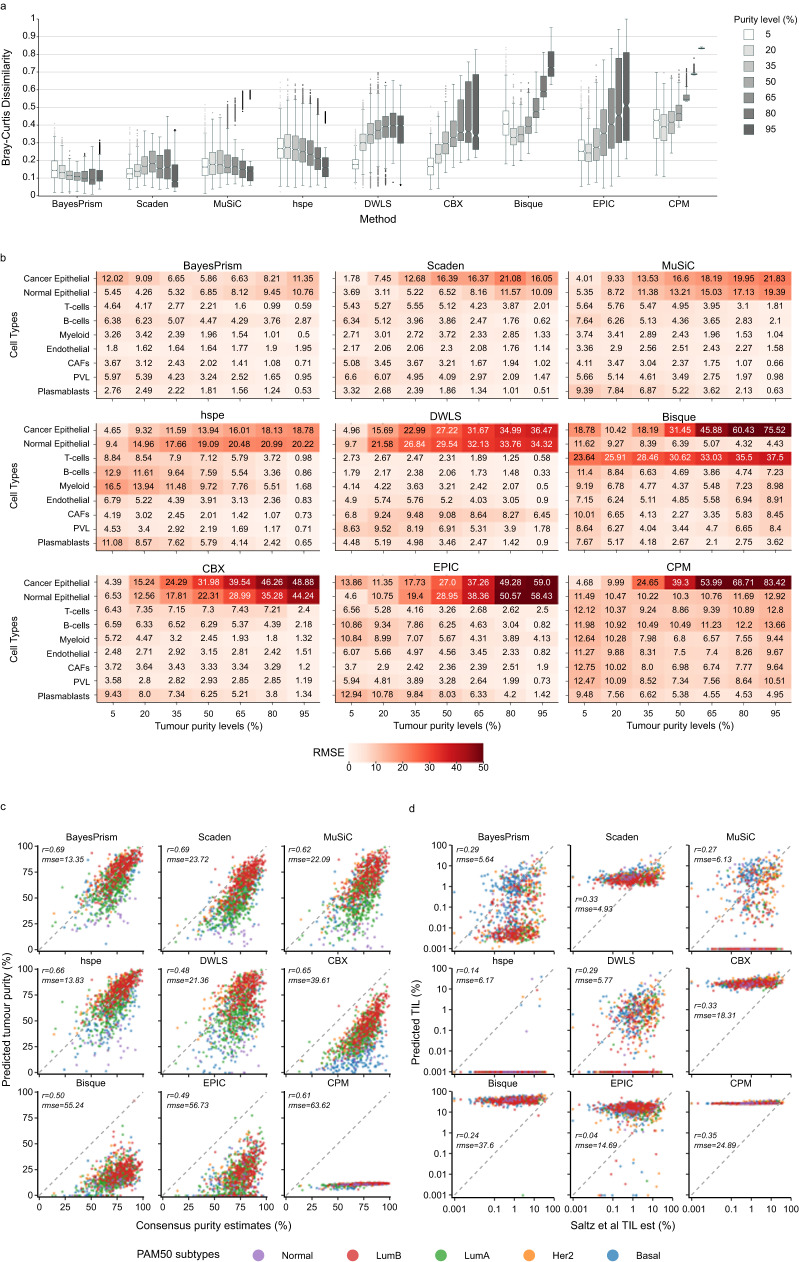
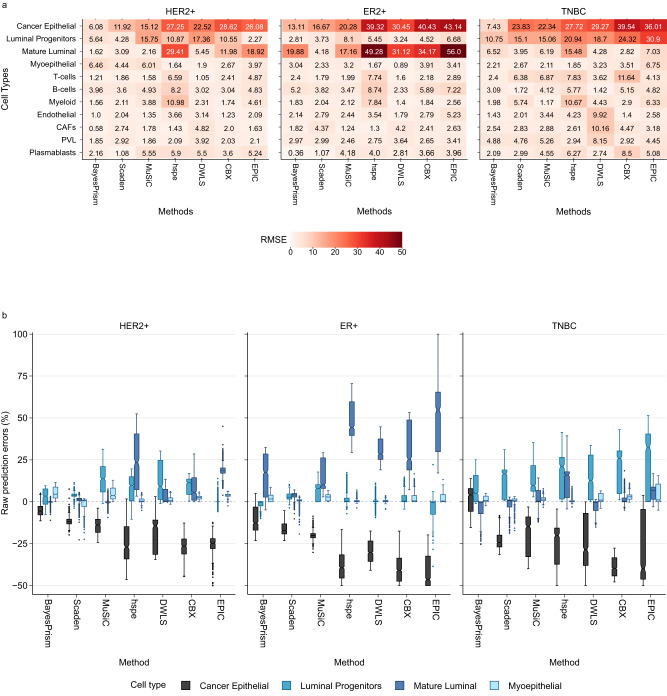
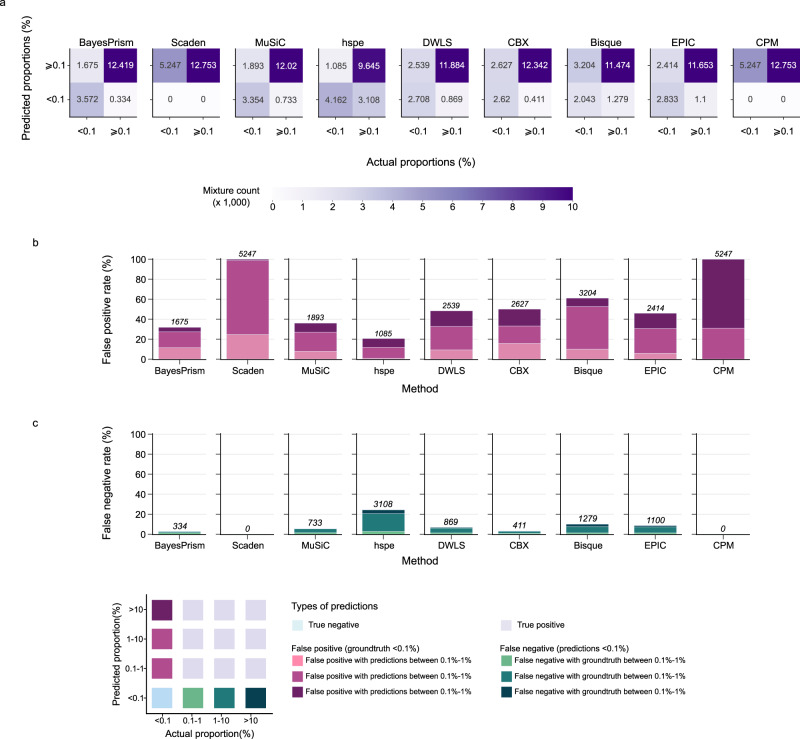
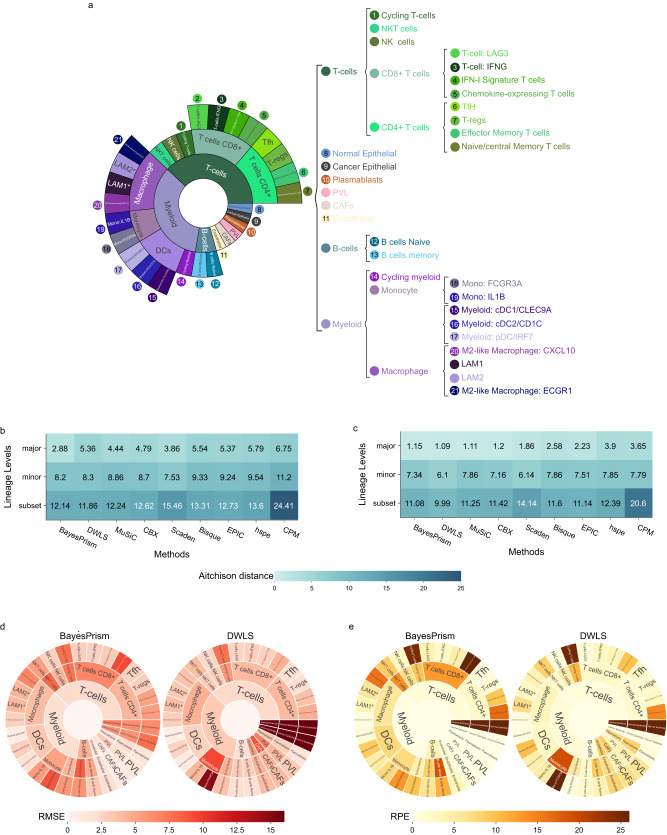
Cells within the tumour microenvironment (TME) can impact tumour development and influence treatment response. Computational approaches have been developed to deconvolve the TME from bulk RNA-seq. Using scRNA-seq profiling from breast tumours we simulate thousands of bulk mixtures, representing tumour purities and cell lineages, to compare the performance of nine TME deconvolution methods (BayesPrism, Scaden, CIBERSORTx, MuSiC, DWLS, hspe, CPM, Bisque, and EPIC). Some methods are more robust in deconvolving mixtures with high tumour purity levels. Most methods tend to mis-predict normal epithelial for cancer epithelial as tumour purity increases, a finding that is validated in two independent datasets. The breast cancer molecular subtype influences this mis-prediction. BayesPrism and DWLS have the lowest combined numbers of false positives and false negatives, and have the best performance when deconvolving granular immune lineages. Our findings highlight the need for more single-cell characterisation of rarer cell types, and suggest that tumour cell compositions should be considered when deconvolving the TME.
© 2023. Springer Nature Limited.
Conflict of interest statement
J.V.P. and N.W. are co-founders of genomiQa. O.K. has consulted for XING Technologies on development of diagnostic assays for HR deficiency. The remaining authors declare that there are no competing interests.
Figures
References
-
- Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J. Cell Sci. 2012;125:5591–5596. - PubMed
-
- Zhang L, et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003;348:203–213. - PubMed
-
- Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017;18:e731–e741. - PubMed
-
- Newell F, et al. Multiomic profiling of checkpoint inhibitor-treated melanoma: Identifying predictors of response and resistance, and markers of biological discordance. Cancer Cell. 2022;40:88–102.e7. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
