

A cleavage rule for selection of increased-fidelity SpCas9 variants with high efficiency and no detectable off-targets
- PMID: 37717069
- PMCID: PMC10505190
- DOI: 10.1038/s41467-023-41393-5
A cleavage rule for selection of increased-fidelity SpCas9 variants with high efficiency and no detectable off-targets
Abstract
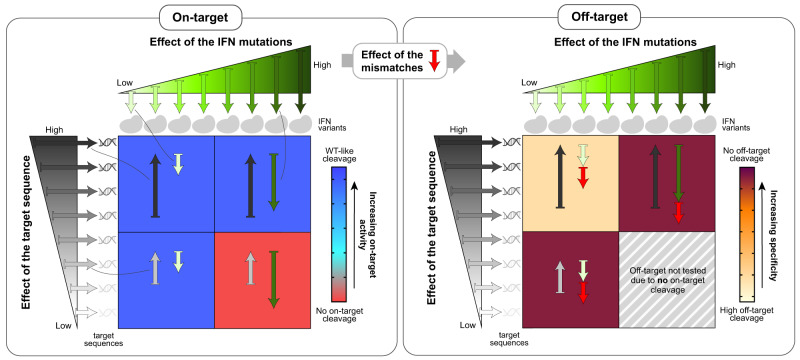
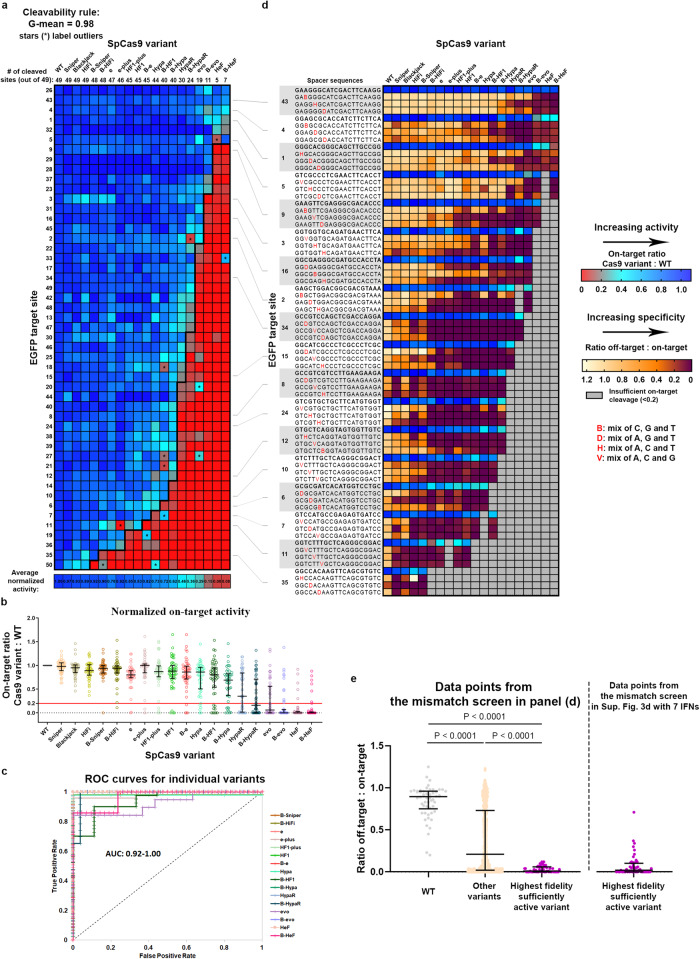
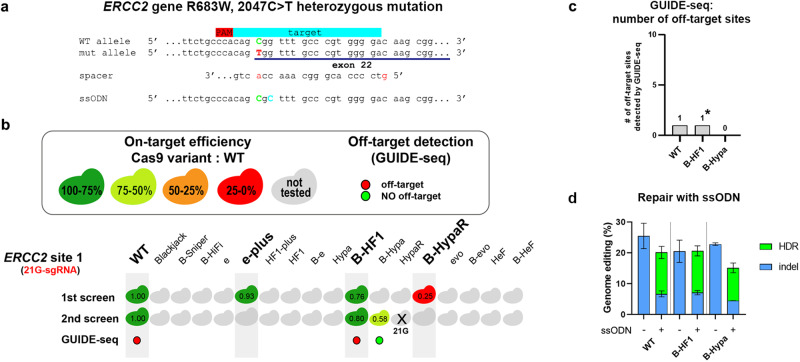
Streptococcus pyogenes Cas9 (SpCas9) has been employed as a genome engineering tool with a promising potential within therapeutics. However, its off-target effects present major safety concerns for applications requiring high specificity. Approaches developed to date to mitigate this effect, including any of the increased-fidelity (i.e., high-fidelity) SpCas9 variants, only provide efficient editing on a relatively small fraction of targets without detectable off-targets. Upon addressing this problem, we reveal a rather unexpected cleavability ranking of target sequences, and a cleavage rule that governs the on-target and off-target cleavage of increased-fidelity SpCas9 variants but not that of SpCas9-NG or xCas9. According to this rule, for each target, an optimal variant with matching fidelity must be identified for efficient cleavage without detectable off-target effects. Based on this insight, we develop here an extended set of variants, the CRISPRecise set, with increased fidelity spanning across a wide range, with differences in fidelity small enough to comprise an optimal variant for each target, regardless of its cleavability ranking. We demonstrate efficient editing with maximum specificity even on those targets that have not been possible in previous studies.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures






Similar articles
-
Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage.Genome Biol. 2017 Oct 6;18(1):190. doi: 10.1186/s13059-017-1318-8. Genome Biol. 2017. PMID: 28985763 Free PMC article.
-
High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects.Nature. 2016 Jan 28;529(7587):490-5. doi: 10.1038/nature16526. Epub 2016 Jan 6. Nature. 2016. PMID: 26735016 Free PMC article.
-
High-throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells.Nat Biomed Eng. 2020 Jan;4(1):111-124. doi: 10.1038/s41551-019-0505-1. Epub 2020 Jan 14. Nat Biomed Eng. 2020. PMID: 31937939
-
In-depth assessment of the PAM compatibility and editing activities of Cas9 variants.Nucleic Acids Res. 2021 Sep 7;49(15):8785-8795. doi: 10.1093/nar/gkab507. Nucleic Acids Res. 2021. PMID: 34133740 Free PMC article.
-
New variants of CRISPR RNA-guided genome editing enzymes.Plant Biotechnol J. 2017 Aug;15(8):917-926. doi: 10.1111/pbi.12736. Epub 2017 May 9. Plant Biotechnol J. 2017. PMID: 28371222 Free PMC article. Review.
Cited by
-
Recent Advances in Tomato Gene Editing.Int J Mol Sci. 2024 Feb 23;25(5):2606. doi: 10.3390/ijms25052606. Int J Mol Sci. 2024. PMID: 38473859 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
