

Epstein-Barr virus-acquired immunodeficiency in myalgic encephalomyelitis-Is it present in long COVID?
- PMID: 37718435
- PMCID: PMC10506247
- DOI: 10.1186/s12967-023-04515-7
Epstein-Barr virus-acquired immunodeficiency in myalgic encephalomyelitis-Is it present in long COVID?
Abstract
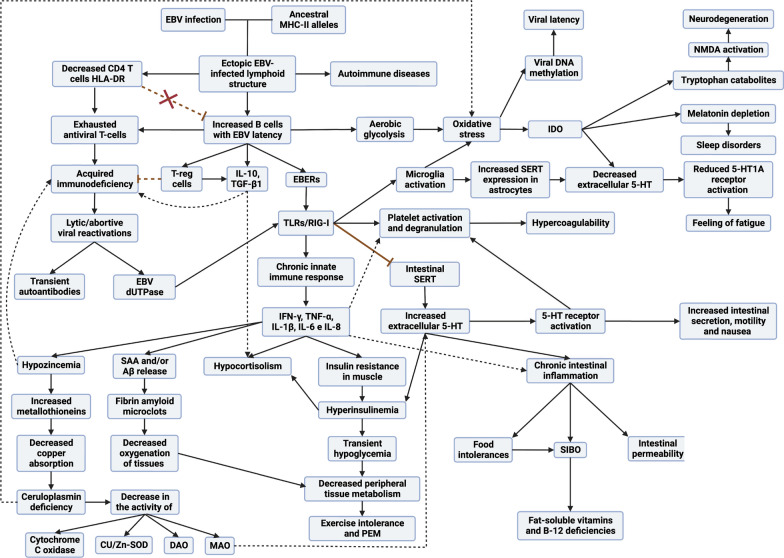
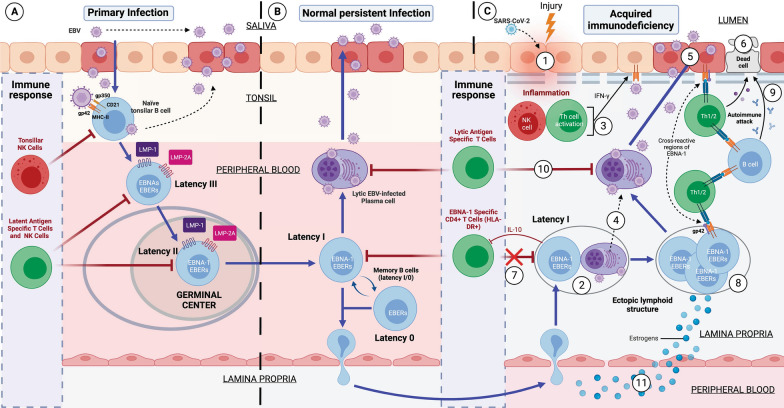
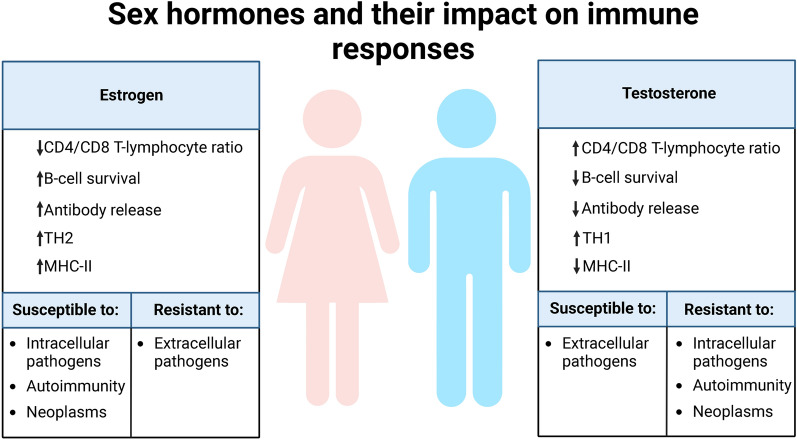
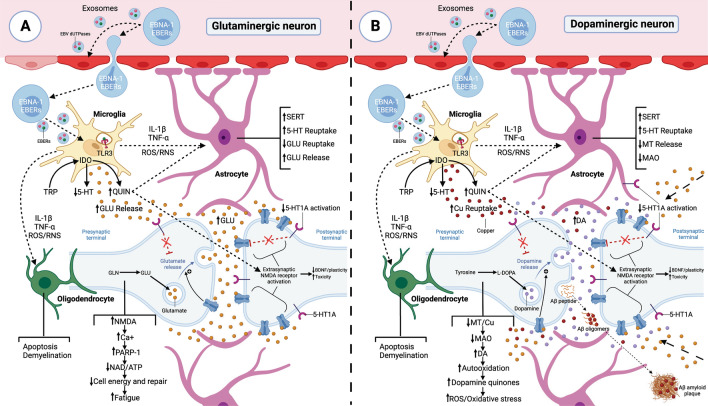
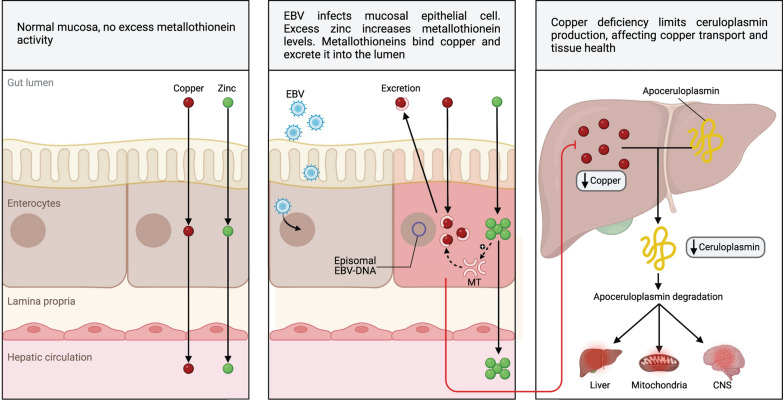
Both myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS) and long COVID (LC) are characterized by similar immunological alterations, persistence of chronic viral infection, autoimmunity, chronic inflammatory state, viral reactivation, hypocortisolism, and microclot formation. They also present with similar symptoms such as asthenia, exercise intolerance, sleep disorders, cognitive dysfunction, and neurological and gastrointestinal complaints. In addition, both pathologies present Epstein-Barr virus (EBV) reactivation, indicating the possibility of this virus being the link between both pathologies. Therefore, we propose that latency and recurrent EBV reactivation could generate an acquired immunodeficiency syndrome in three steps: first, an acquired EBV immunodeficiency develops in individuals with "weak" EBV HLA-II haplotypes, which prevents the control of latency I cells. Second, ectopic lymphoid structures with EBV latency form in different tissues (including the CNS), promoting inflammatory responses and further impairment of cell-mediated immunity. Finally, immune exhaustion occurs due to chronic exposure to viral antigens, with consolidation of the disease. In the case of LC, prior to the first step, there is the possibility of previous SARS-CoV-2 infection in individuals with "weak" HLA-II haplotypes against this virus and/or EBV.
Keywords: Chronic fatigue syndrome; EBV EBNA-1; HLA-II alleles; Immunodeficiency; Inflammation; Long COVID syndrome; Myalgic encephalomyelitis; Post-acute COVID-19 syndrome.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Persistent SARS-CoV-2 Infection, EBV, HHV-6 and Other Factors May Contribute to Inflammation and Autoimmunity in Long COVID.Viruses. 2023 Jan 31;15(2):400. doi: 10.3390/v15020400. Viruses. 2023. PMID: 36851614 Free PMC article. Review.
-
Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome.Front Immunol. 2021 Nov 15;12:656797. doi: 10.3389/fimmu.2021.656797. eCollection 2021. Front Immunol. 2021. PMID: 34867935 Free PMC article. Review.
-
Saliva antibody-fingerprint of reactivated latent viruses after mild/asymptomatic COVID-19 is unique in patients with myalgic-encephalomyelitis/chronic fatigue syndrome.Front Immunol. 2022 Oct 20;13:949787. doi: 10.3389/fimmu.2022.949787. eCollection 2022. Front Immunol. 2022. PMID: 36341457 Free PMC article.
-
Early Growth Response Gene Upregulation in Epstein-Barr Virus (EBV)-Associated Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS).Biomolecules. 2020 Oct 26;10(11):1484. doi: 10.3390/biom10111484. Biomolecules. 2020. PMID: 33114612 Free PMC article. Review.
-
Incidence of Epstein-Barr virus reactivation is elevated in COVID-19 patients.Virus Res. 2023 Sep;334:199157. doi: 10.1016/j.virusres.2023.199157. Epub 2023 Jun 26. Virus Res. 2023. PMID: 37364815 Free PMC article.
Cited by
-
Human herpesvirus reactivation and its potential role in the pathogenesis of post-acute sequelae of SARS-CoV-2 infection.Geroscience. 2025 Feb;47(1):167-187. doi: 10.1007/s11357-024-01323-9. Epub 2024 Aug 29. Geroscience. 2025. PMID: 39207648 Free PMC article. Review.
-
Cerebromicrovascular mechanisms contributing to long COVID: implications for neurocognitive health.Geroscience. 2025 Feb;47(1):745-779. doi: 10.1007/s11357-024-01487-4. Epub 2025 Jan 7. Geroscience. 2025. PMID: 39777702 Free PMC article. Review.
-
HERV Dysregulation in a Case of Myalgic Encephalomyelitis and Multiple Sclerosis Responsive to Rituximab.Int J Mol Sci. 2025 May 20;26(10):4885. doi: 10.3390/ijms26104885. Int J Mol Sci. 2025. PMID: 40430026 Free PMC article.
-
Hypocortisolemic ASIA: a vaccine- and chronic infection-induced syndrome behind the origin of long COVID and myalgic encephalomyelitis.Front Immunol. 2024 Jul 9;15:1422940. doi: 10.3389/fimmu.2024.1422940. eCollection 2024. Front Immunol. 2024. PMID: 39044822 Free PMC article. Review.
-
Possible Role of Cannabis in the Management of Neuroinflammation in Patients with Post-COVID Condition.Int J Mol Sci. 2024 Mar 29;25(7):3805. doi: 10.3390/ijms25073805. Int J Mol Sci. 2024. PMID: 38612615 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
