

Computational investigation of unsaturated ketone derivatives as MAO-B inhibitors by using QSAR, ADME/Tox, molecular docking, and molecular dynamics simulations
- PMID: 37720619
- PMCID: PMC10503977
- DOI: 10.55730/1300-0527.3360
Computational investigation of unsaturated ketone derivatives as MAO-B inhibitors by using QSAR, ADME/Tox, molecular docking, and molecular dynamics simulations
Abstract
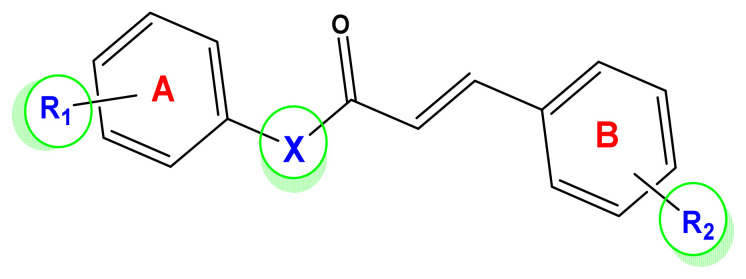
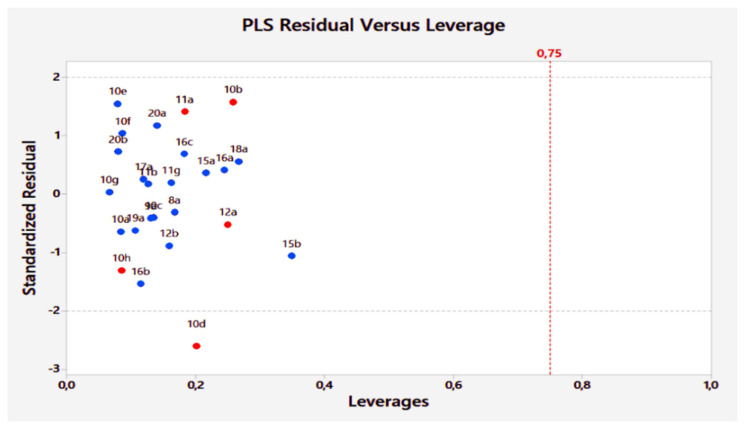
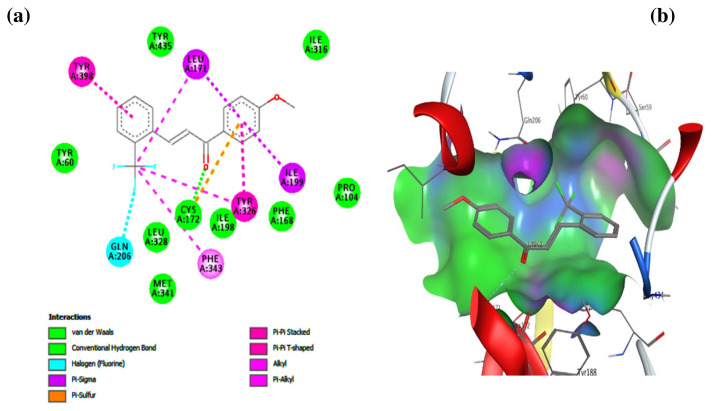
Unsaturated ketone derivatives are known as monoamine oxidase B (MAO-B) inhibitors, a potential drug target for Parkinson's disease. Here, molecular modeling studies, including 2D-QSAR, ADMET prediction, molecular docking, and MD simulation, were performed on a new series of MAO-B inhibitors. The objective is to identify new MAO-B inhibitors with high inhibitory efficacy. The developed 2D-QSAR model was based on the descriptors of MOE software. The most appropriate model, using the partial least squares regression (PLS regression) method, yielded 0.88 for the determination coefficient (r2), 0.28 for the root-mean-square error (RMSE), and 0.2 for the mean absolute error (MAE). The predictive capacity of the generated model was evaluated by internal and external validations, which gave the Q2 and R2test values of 0.81 and 0.71, respectively. The ability of a compound to be orally active was determined using the drug-likeness and ADMET prediction. The results indicate that most of the compounds have moderate pharmacokinetic characteristics without any side effects. Furthermore, the affinity of the ligands (unsaturated ketone derivatives) to the MAO-B receptor was determined using molecular docking. The top conformers were then subjected to MD simulation. This research may pave the way for the development of novel unsaturated ketone derivatives capable of inhibiting the MAO-B enzyme.
Keywords: MAO-B inhibitors; Parkinson’s disease; ligand-based drug design; structure-based drug design.
© TÜBİTAK.
Conflict of interest statement
Conflict of interest All authors declare that they have no conflict of interest in this work.
Figures







References
-
- Jegham S, George P, Recherche S, Snc DDR, Carrières R. Monoamine oxidase A and B inhibitors. Expert Opinion on Therapeutic Patents Jegham. 1998;8(9):1143–1150. doi: 10.1517/13543776.8.9.1143. - DOI
LinkOut - more resources
Full Text Sources