

Clinical and genetic characterisation of a large Indian congenital myasthenic syndrome cohort
- PMID: 37721175
- PMCID: PMC10766255
- DOI: 10.1093/brain/awad315
Clinical and genetic characterisation of a large Indian congenital myasthenic syndrome cohort
Abstract
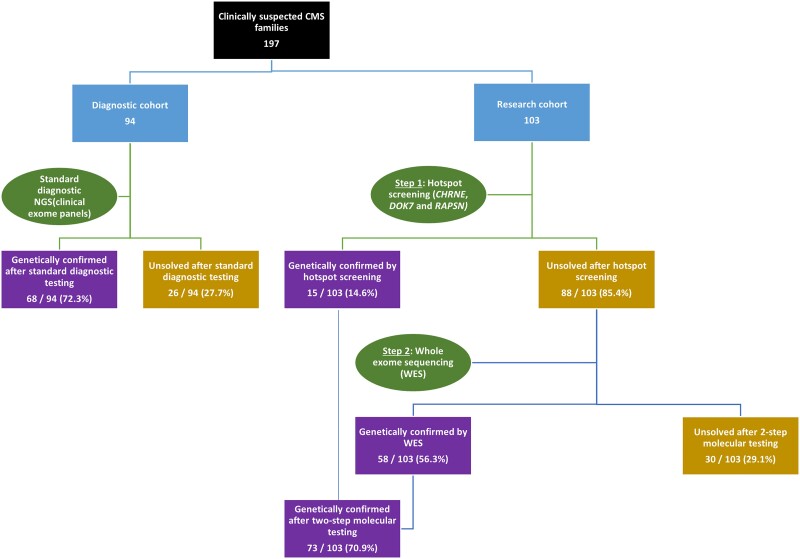
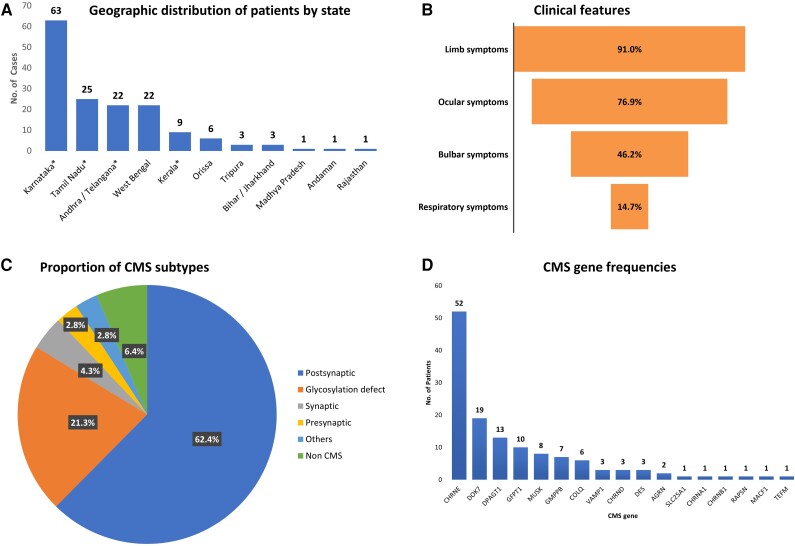
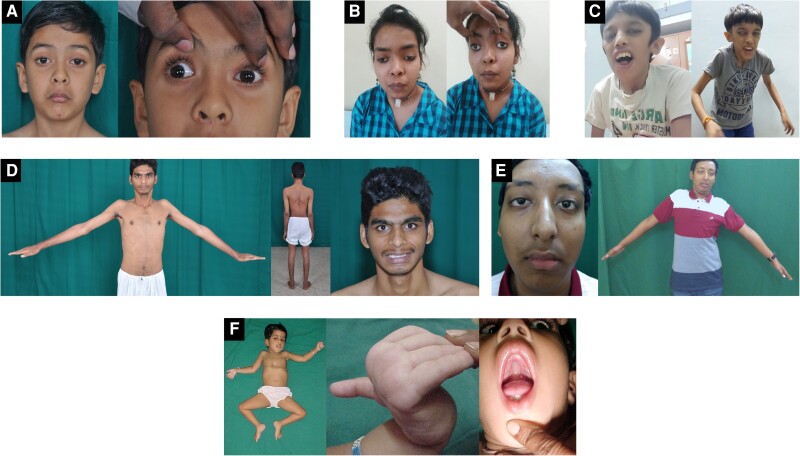
Congenital myasthenic syndromes (CMS) are a rare group of inherited disorders caused by gene defects associated with the neuromuscular junction and potentially treatable with commonly available medications such as acetylcholinesterase inhibitors and β2 adrenergic receptor agonists. In this study, we identified and genetically characterized the largest cohort of CMS patients from India to date. Genetic testing of clinically suspected patients evaluated in a South Indian hospital during the period 2014-19 was carried out by standard diagnostic gene panel testing or using a two-step method that included hotspot screening followed by whole-exome sequencing. In total, 156 genetically diagnosed patients (141 families) were characterized and the mutational spectrum and genotype-phenotype correlation described. Overall, 87 males and 69 females were evaluated, with the age of onset ranging from congenital to fourth decade (mean 6.6 ± 9.8 years). The mean age at diagnosis was 19 ± 12.8 (1-56 years), with a mean diagnostic delay of 12.5 ± 9.9 (0-49 years). Disease-causing variants in 17 CMS-associated genes were identified in 132 families (93.6%), while in nine families (6.4%), variants in genes not associated with CMS were found. Overall, postsynaptic defects were most common (62.4%), followed by glycosylation defects (21.3%), synaptic basal lamina genes (4.3%) and presynaptic defects (2.8%). Other genes found to cause neuromuscular junction defects (DES, TEFM) in our cohort accounted for 2.8%. Among the individual CMS genes, the most commonly affected gene was CHRNE (39.4%), followed by DOK7 (14.4%), DPAGT1 (9.8%), GFPT1 (7.6%), MUSK (6.1%), GMPPB (5.3%) and COLQ (4.5%). We identified 22 recurrent variants in this study, out of which eight were found to be geographically specific to the Indian subcontinent. Apart from the known common CHRNE variants p.E443Kfs*64 (11.4%) and DOK7 p.A378Sfs*30 (9.3%), we identified seven novel recurrent variants specific to this cohort, including DPAGT1 p.T380I and DES c.1023+5G>A, for which founder haplotypes are suspected. This study highlights the geographic differences in the frequencies of various causative CMS genes and underlines the increasing significance of glycosylation genes (DPAGT1, GFPT1 and GMPPB) as a cause of neuromuscular junction defects. Myopathy and muscular dystrophy genes such as GMPPB and DES, presenting as gradually progressive limb girdle CMS, expand the phenotypic spectrum. The novel genes MACF1 and TEFM identified in this cohort add to the expanding list of genes with new mechanisms causing neuromuscular junction defects.
Keywords: NGS; congenital myasthenic syndromes; genetics; neuromuscular junction; recurrent mutations.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
The authors report no competing interests.
Figures
References
-
- McMacken G, Abicht A, Evangelista T, Spendiff S, Lochmüller H. The increasing genetic and phenotypical diversity of congenital myasthenic syndromes. Neuropediatrics. 2017;48:294–308. - PubMed
-
- Ramdas S, Beeson D. Congenital myasthenic syndromes: Where do we go from here? Neuromuscul Disord. 2021;31:943–954. - PubMed
-
- Parr JR, Andrew MJ, Finnis M, Beeson D, Vincent A, Jayawant S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child. 2014;99:539–542. - PubMed
-
- Natera-de Benito D, Töpf A, Vilchez JJ, et al. . Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul Disord. 2017;27:1087–1098. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
