

Microglia moonlighting after traumatic brain injury: aging and interferons influence chronic microglia reactivity
- PMID: 37723009
- PMCID: PMC10592045
- DOI: 10.1016/j.tins.2023.08.008
Microglia moonlighting after traumatic brain injury: aging and interferons influence chronic microglia reactivity
Abstract
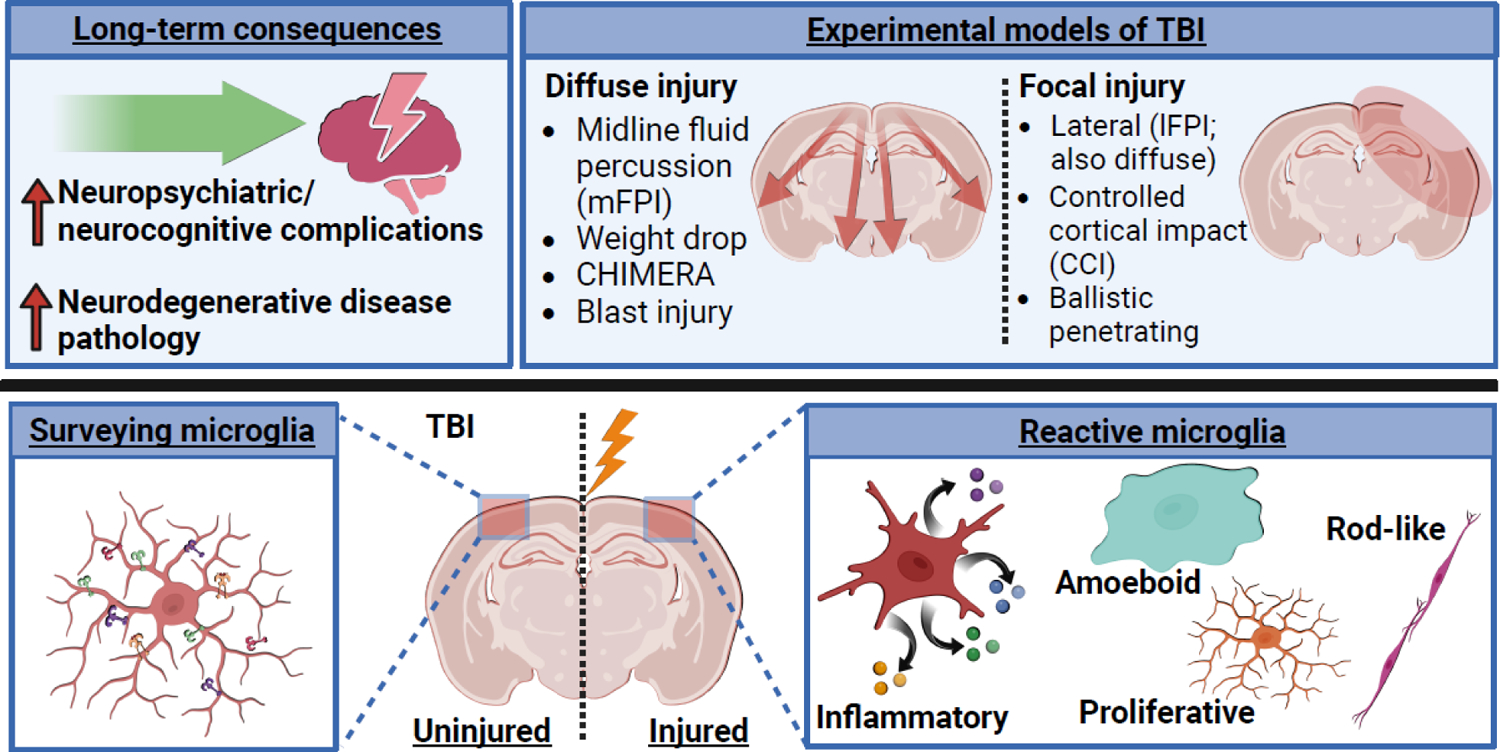
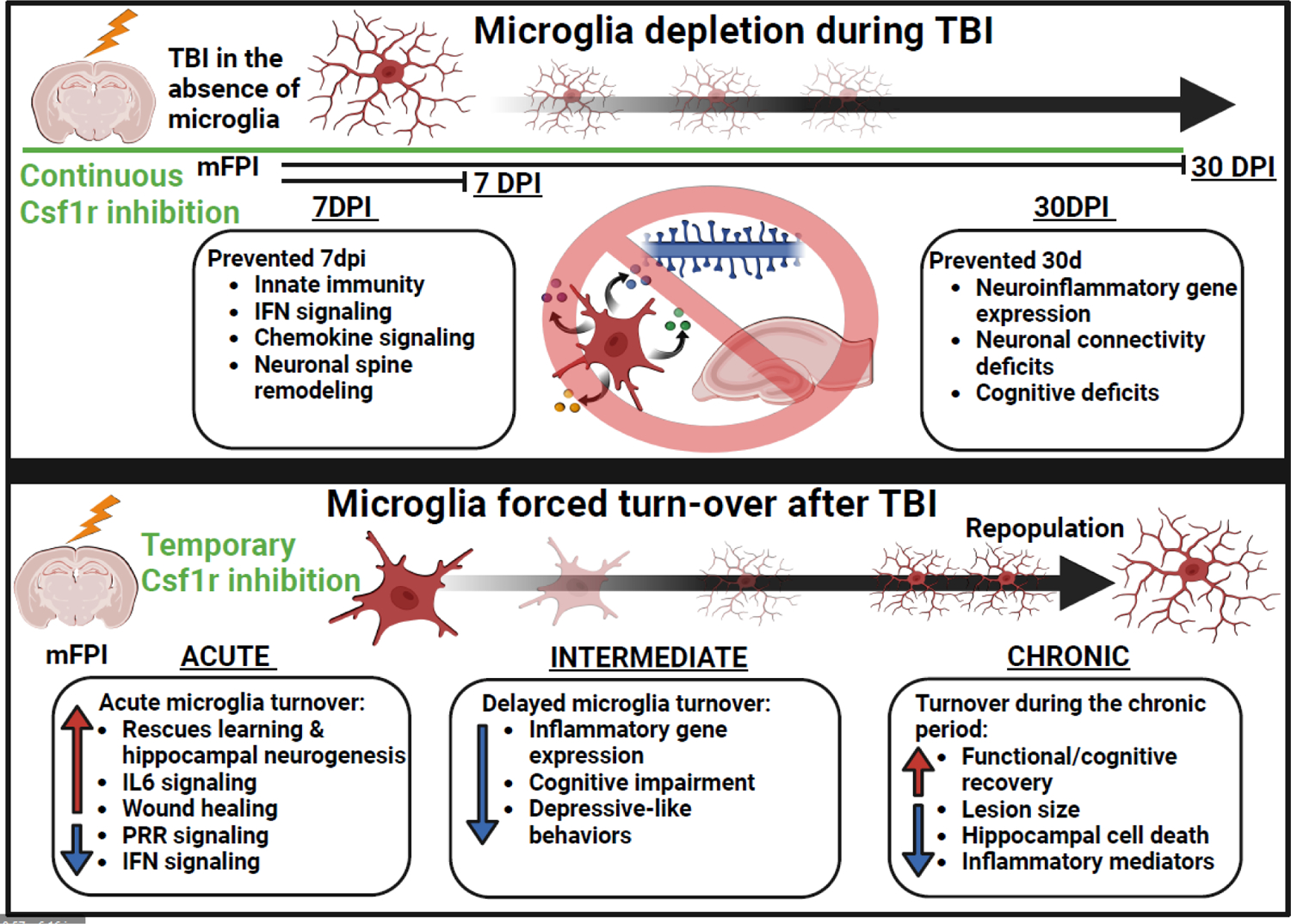
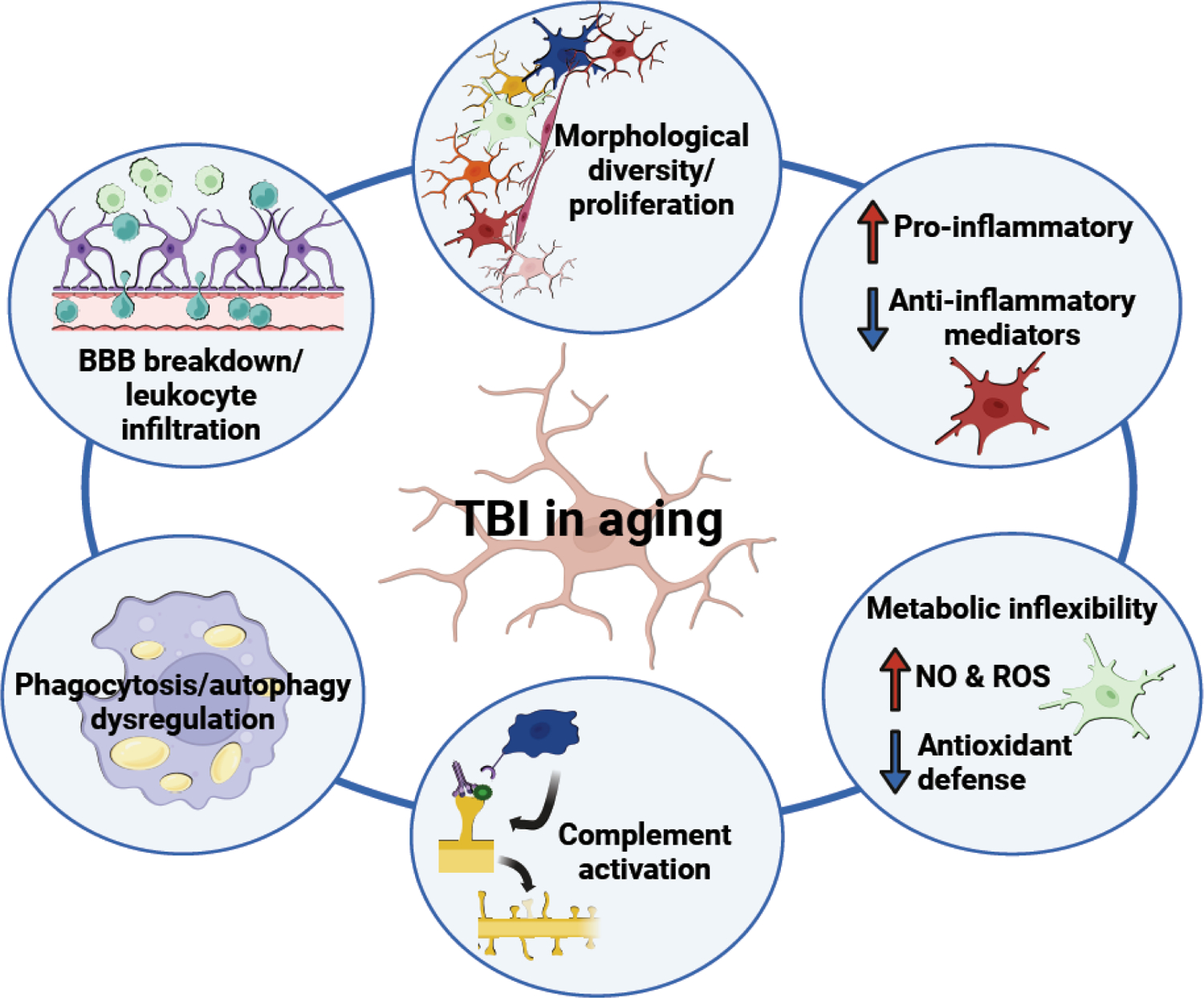
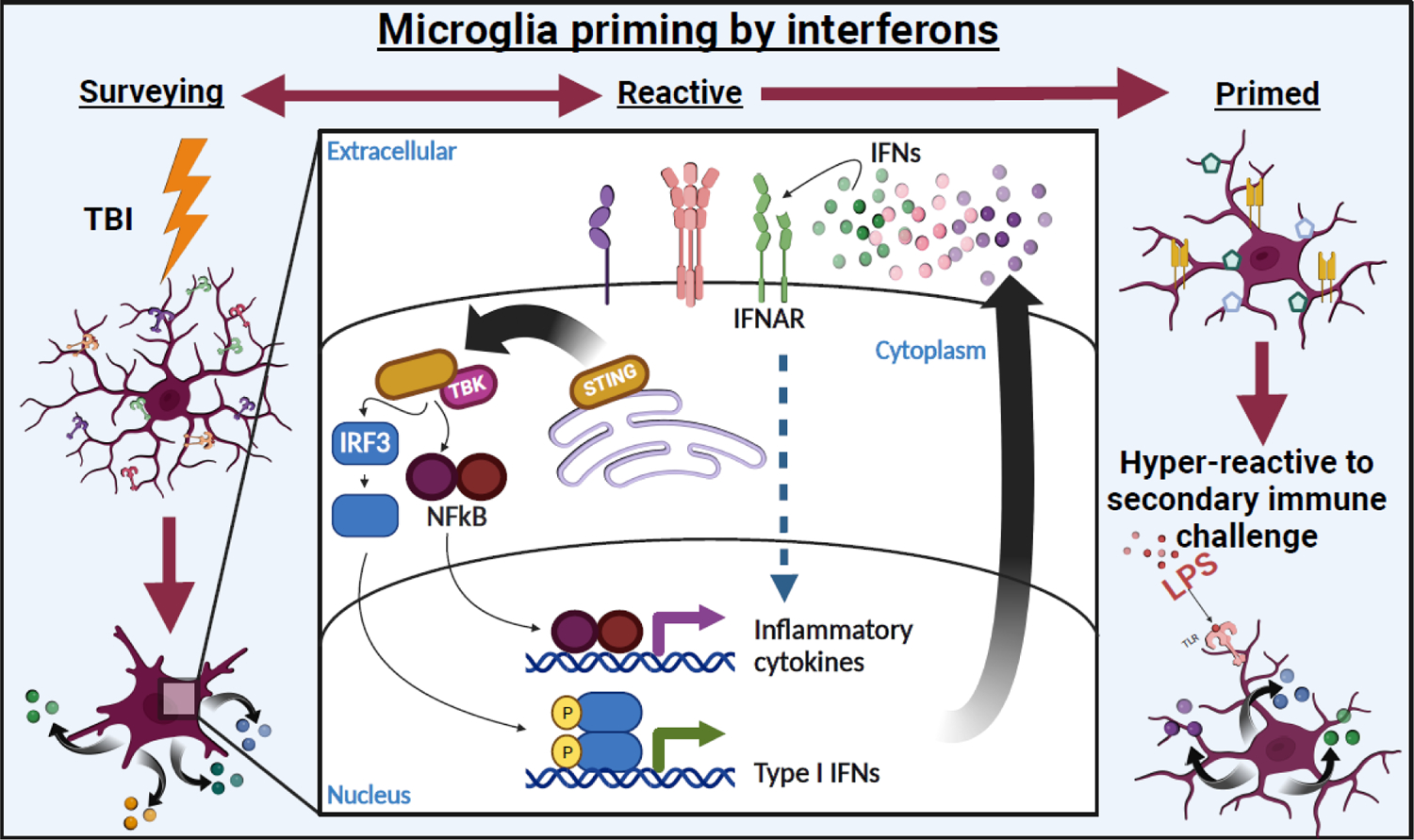
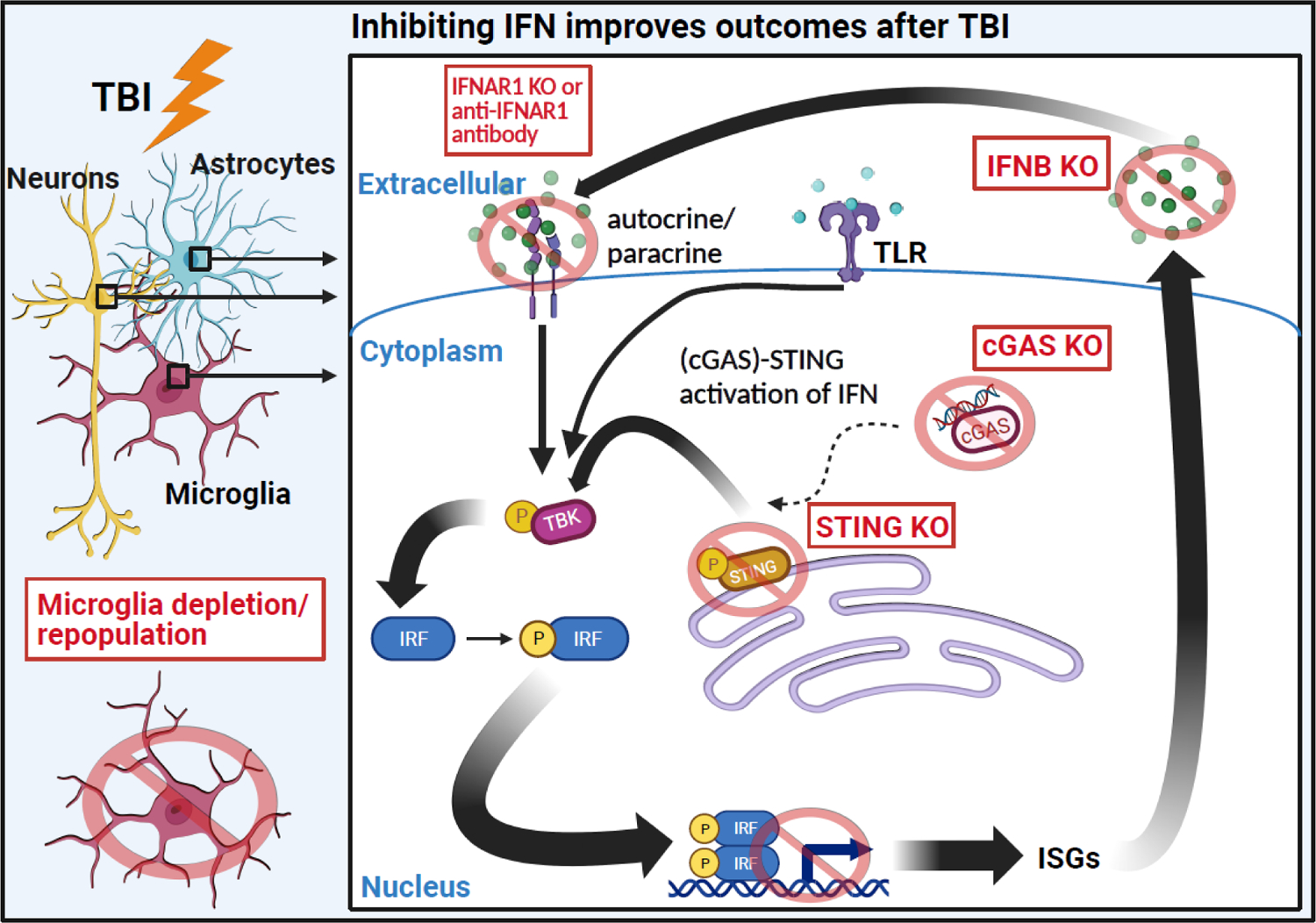
Most of the individuals who experience traumatic brain injury (TBI) develop neuropsychiatric and cognitive complications that negatively affect recovery and health span. Activation of multiple inflammatory pathways persists after TBI, but it is unclear how inflammation contributes to long-term behavioral and cognitive deficits. One outcome of TBI is microglial priming and subsequent hyper-reactivity to secondary stressors, injuries, or immune challenges that further augment complications. Additionally, microglia priming with aging contributes to exaggerated glial responses to TBI. One prominent inflammatory pathway, interferon (IFN) signaling, is increased after TBI and may contribute to microglial priming and subsequent reactivity. This review discusses the contributions of microglia to inflammatory processes after TBI, as well as the influence of aging and IFNs on microglia reactivity and chronic inflammation after TBI.
Keywords: microglia priming; neurodegeneration; neuroinflammation; neurotrauma.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests in relation to this work.
Figures
References
-
- Gualtieri T and Cox DR, The delayed neurobehavioural sequelae of traumatic brain injury. Brain Injury, 1991. 5(3): p. 219–232. - PubMed
-
- Millis SR, et al. , Long-term neuropsychological outcome after traumatic brain injury. J Head Trauma Rehabil, 2001. 16(4): p. 343–55. - PubMed
-
- Salmond CH, et al. , Changes over time in cognitive and structural profiles of head injury survivors. Neuropsychologia, 2006. 44(10): p. 1995–1998. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
