

Whole-genome screens reveal regulators of differentiation state and context-dependent migration in human neutrophils
- PMID: 37723145
- PMCID: PMC10507112
- DOI: 10.1038/s41467-023-41452-x
Whole-genome screens reveal regulators of differentiation state and context-dependent migration in human neutrophils
Abstract
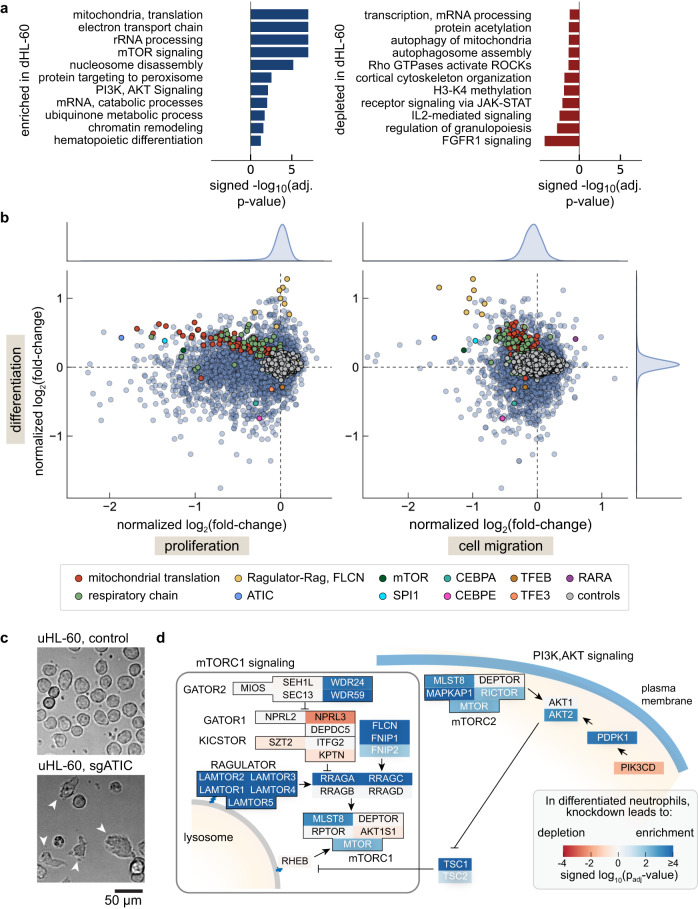
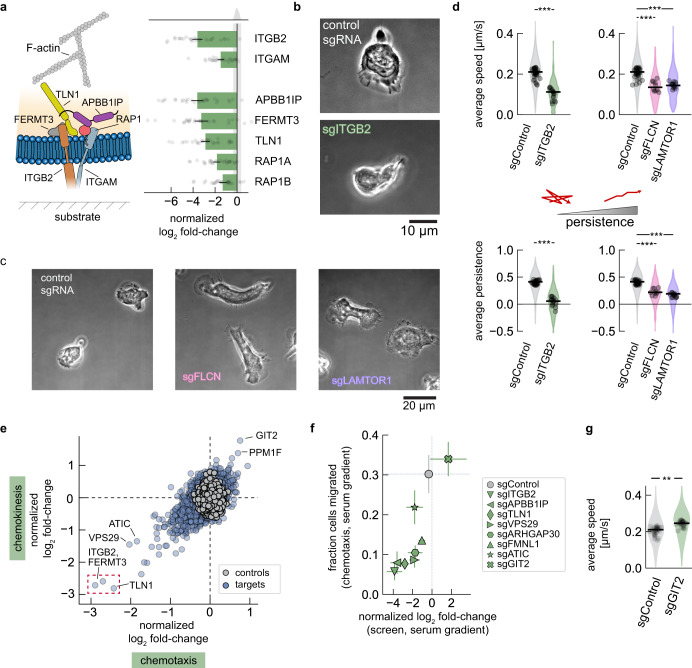
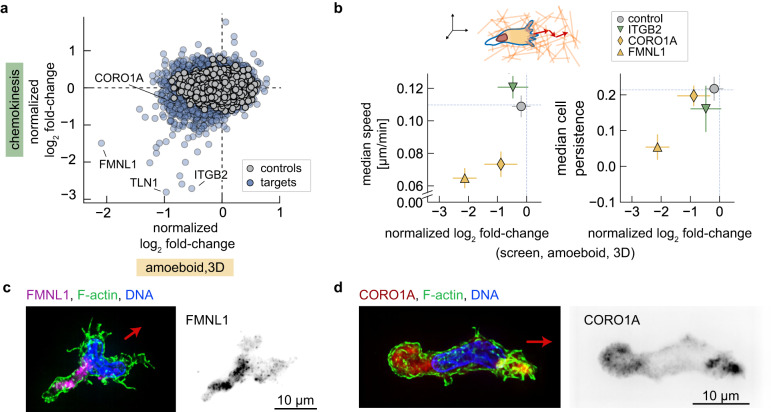
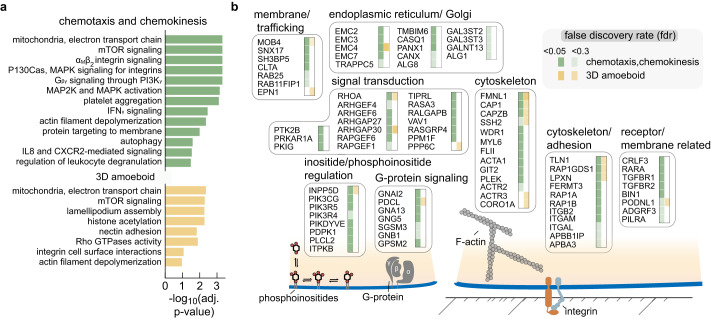
Neutrophils are the most abundant leukocyte in humans and provide a critical early line of defense as part of our innate immune system. We perform a comprehensive, genome-wide assessment of the molecular factors critical to proliferation, differentiation, and cell migration in a neutrophil-like cell line. Through the development of multiple migration screen strategies, we specifically probe directed (chemotaxis), undirected (chemokinesis), and 3D amoeboid cell migration in these fast-moving cells. We identify a role for mTORC1 signaling in cell differentiation, which influences neutrophil abundance, survival, and migratory behavior. Across our individual migration screens, we identify genes involved in adhesion-dependent and adhesion-independent cell migration, protein trafficking, and regulation of the actomyosin cytoskeleton. This genome-wide screening strategy, therefore, provides an invaluable approach to the study of neutrophils and provides a resource that will inform future studies of cell migration in these and other rapidly migrating cells.
© 2023. Springer Nature Limited.
Conflict of interest statement
J.A.T. is chief scientific advisor at the Allen Institute for Cell Science (Seattle, WA, 98109). The authors otherwise declare no competing interests.
Figures




References
-
- Sender R, Milo R. The distribution of cellular turnover in the human body. Nat. Med. 2021;27:45–48. - PubMed
-
- Evrard M, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. 2018;48:364–379.e8. - PubMed
-
- Liew PX, Kubes P. The neutrophil’s role during health and disease. Physiol. Rev. 2019;99:1223–1248. - PubMed
-
- Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020;19:253–275. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
