

Loss of Grin2a causes a transient delay in the electrophysiological maturation of hippocampal parvalbumin interneurons
- PMID: 37723282
- PMCID: PMC10507040
- DOI: 10.1038/s42003-023-05298-9
Loss of Grin2a causes a transient delay in the electrophysiological maturation of hippocampal parvalbumin interneurons
Abstract
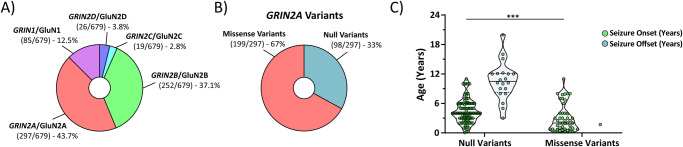
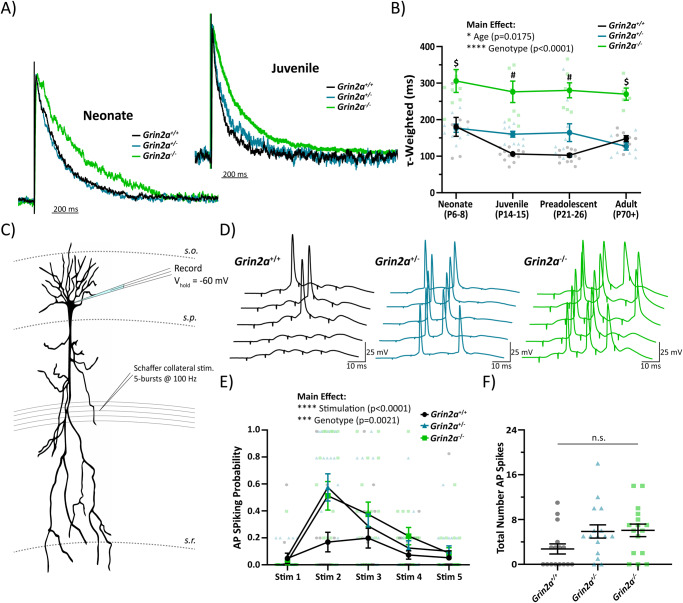
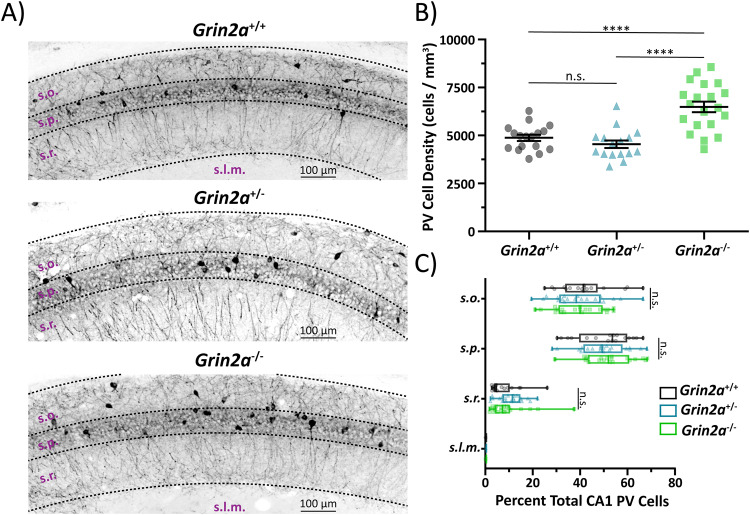
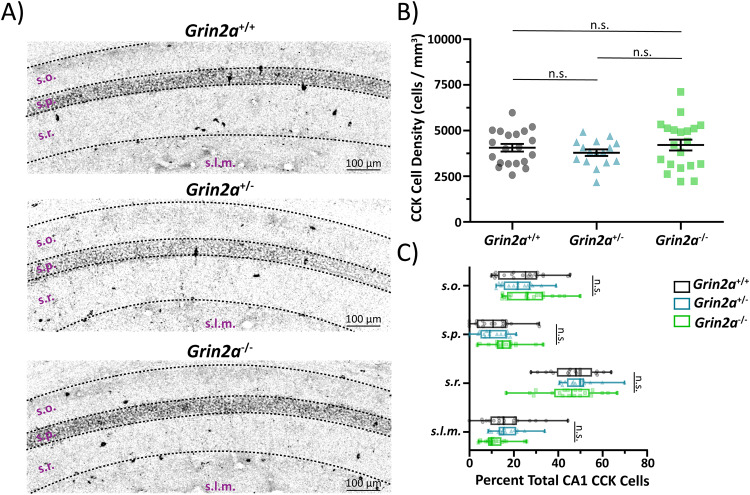
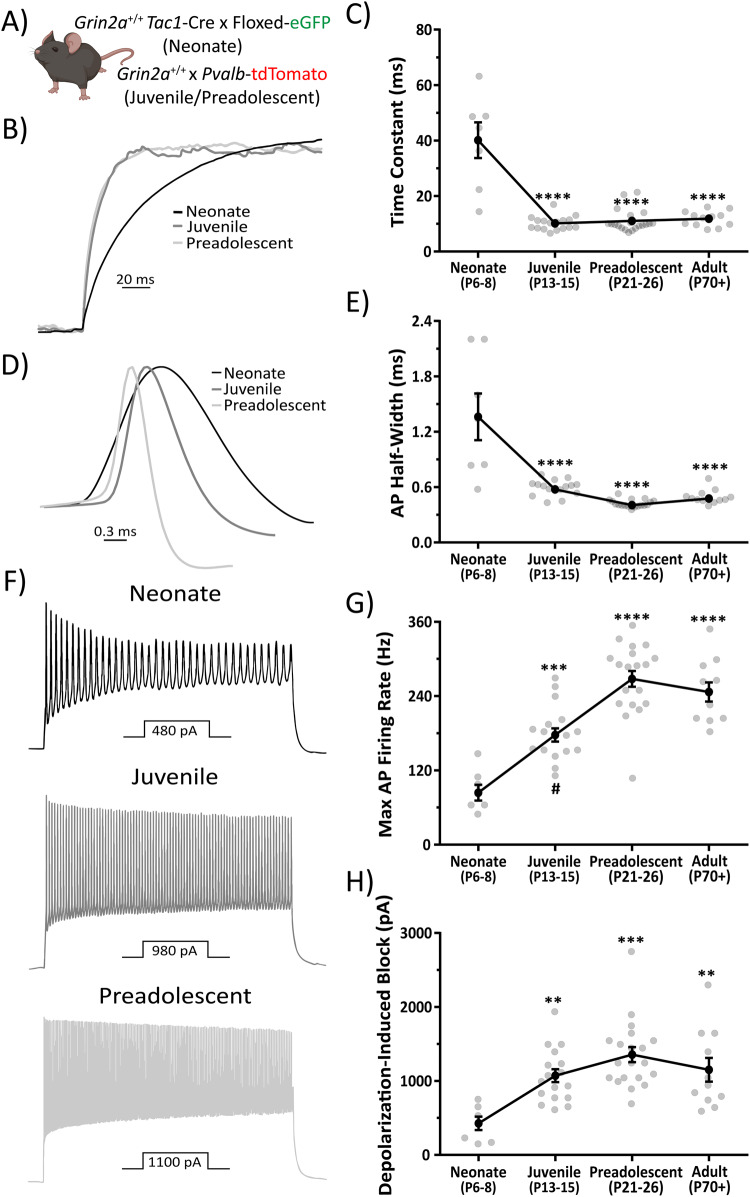
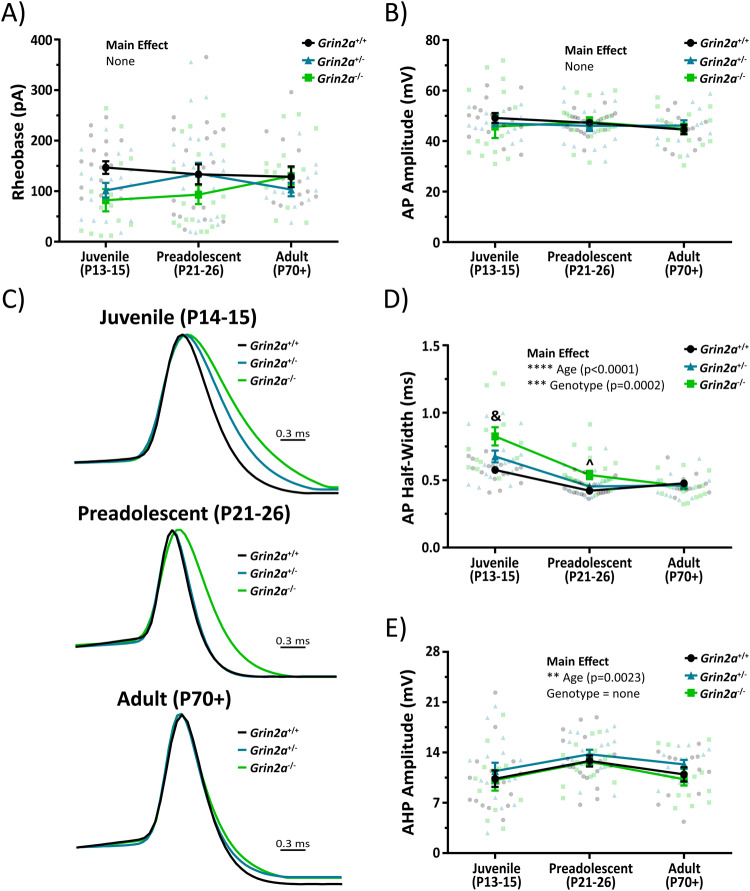
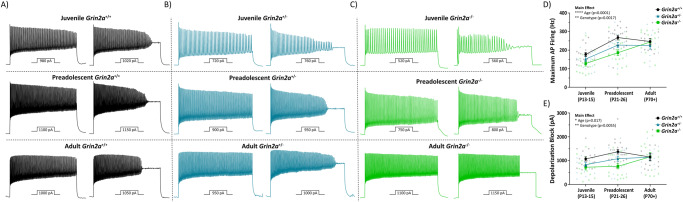
N-methyl-D-aspartate receptors (NMDARs) are ligand-gated ionotropic glutamate receptors that mediate a calcium-permeable component to fast excitatory neurotransmission. NMDARs are heterotetrameric assemblies of two obligate GluN1 subunits (GRIN1) and two GluN2 subunits (GRIN2A-GRIN2D). Sequencing data shows that 43% (297/679) of all currently known NMDAR disease-associated genetic variants are within the GRIN2A gene, which encodes the GluN2A subunit. Here, we show that unlike missense GRIN2A variants, individuals affected with disease-associated null GRIN2A variants demonstrate a transient period of seizure susceptibility that begins during infancy and diminishes near adolescence. We show increased circuit excitability and CA1 pyramidal cell output in juvenile mice of both Grin2a+/- and Grin2a-/- mice. These alterations in somatic spiking are not due to global upregulation of most Grin genes (including Grin2b). Deeper evaluation of the developing CA1 circuit led us to uncover age- and Grin2a gene dosing-dependent transient delays in the electrophysiological maturation programs of parvalbumin (PV) interneurons. We report that Grin2a+/+ mice reach PV cell electrophysiological maturation between the neonatal and juvenile neurodevelopmental timepoints, with Grin2a+/- mice not reaching PV cell electrophysiological maturation until preadolescence, and Grin2a-/- mice not reaching PV cell electrophysiological maturation until adulthood. Overall, these data may represent a molecular mechanism describing the transient nature of seizure susceptibility in disease-associated null GRIN2A patients.
© 2023. Springer Nature Limited.
Conflict of interest statement
H.Y. and S.F.T. are co-inventors of Emory-owned intellectual property. S.F.T. is a member of the SAB for Sage Therapeutics, Eumentis Therapeutics, the GRIN2B Foundation, the CureGRIN Foundation, and CombinedBrain. S.F.T. is a consultant for GRIN Therapeutics and Neurocrine. H.Y. is the PI on a research grant from Sage Therapeutics to Emory. S.F.T. is cofounder of NeurOp, Inc. and Agrithera. T.A.B. is a member of the SAB for GRIN2B Foundation, CureGRIN Foundation, GRIN Therapeutics, and Neurocrine; all remuneration has been made to his department. T.G.B. is PI on a research grant from Neumora to Emory University. J.R.L. is a consultant for GRIN Therapeutics and a member of the SAB for GRIN2B Foundation and CureGRIN Foundation. All other authors declare no competing interests.
Figures

References
-
- Collingridge G. Synaptic plasticity. The role of NMDA receptors in learning and memory. Nature. 1987;330:604–605. - PubMed
-
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat. Rev. Neurosci. 2004;5:361–372. - PubMed
-
- Watanabe D, et al. Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell. 1998;95:17–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
