Identification of novel genetic risk factors of dilated cardiomyopathy: from canine to human
- PMID: 37723491
- PMCID: PMC10506233
- DOI: 10.1186/s13073-023-01221-3
Identification of novel genetic risk factors of dilated cardiomyopathy: from canine to human
Abstract
Background: Dilated cardiomyopathy (DCM) is a life-threatening heart disease and a common cause of heart failure due to systolic dysfunction and subsequent left or biventricular dilatation. A significant number of cases have a genetic etiology; however, as a complex disease, the exact genetic risk factors are largely unknown, and many patients remain without a molecular diagnosis.
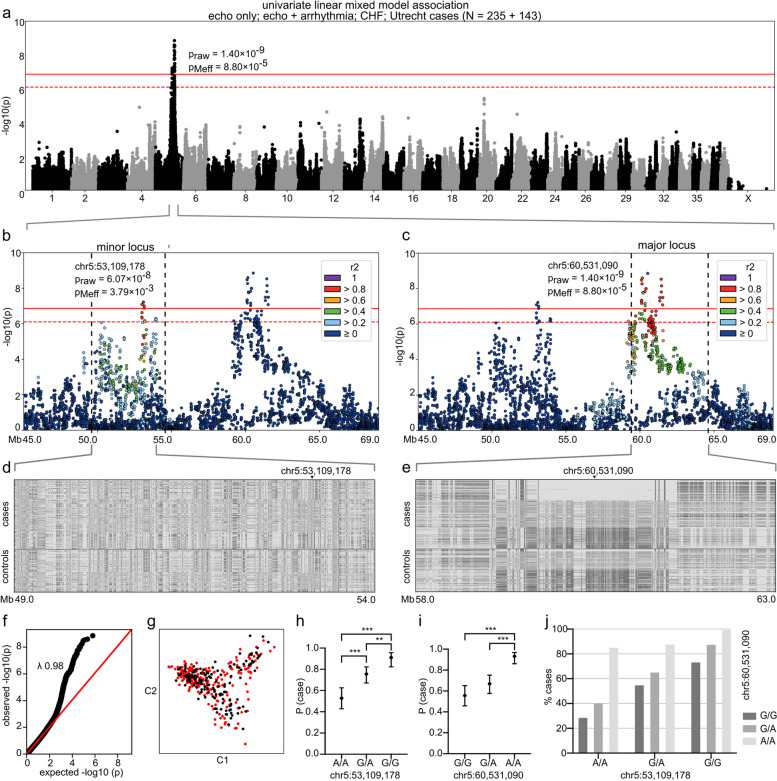
Methods: We performed GWAS followed by whole-genome, transcriptome, and immunohistochemical analyses in a spontaneously occurring canine model of DCM. Canine gene discovery was followed up in three human DCM cohorts.
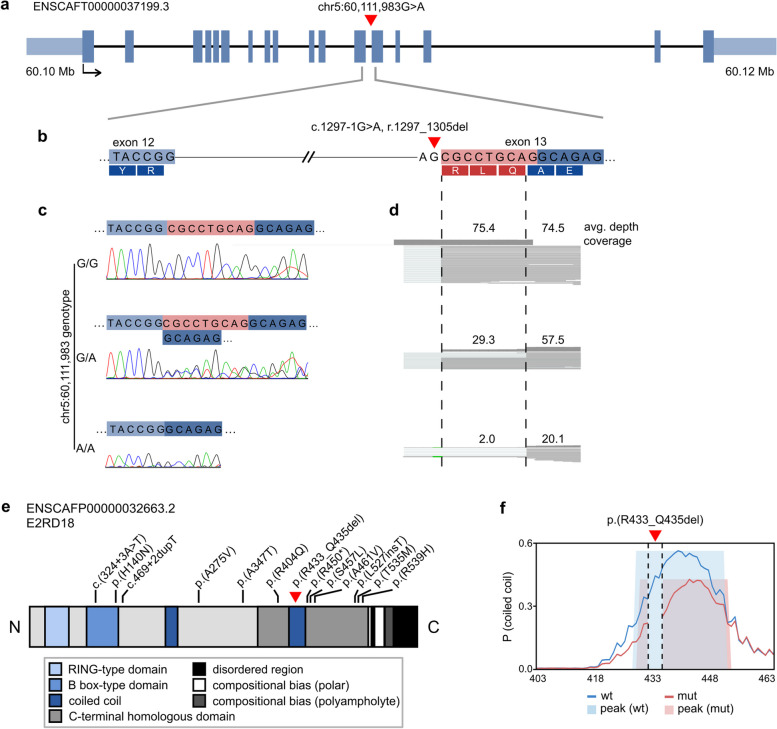
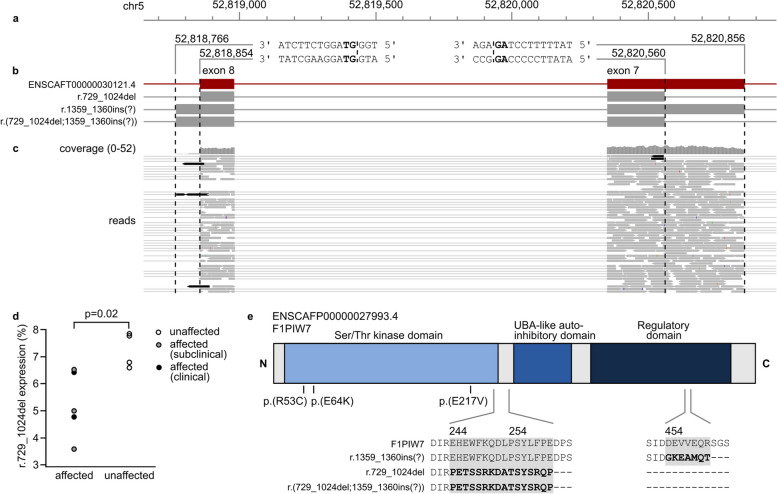
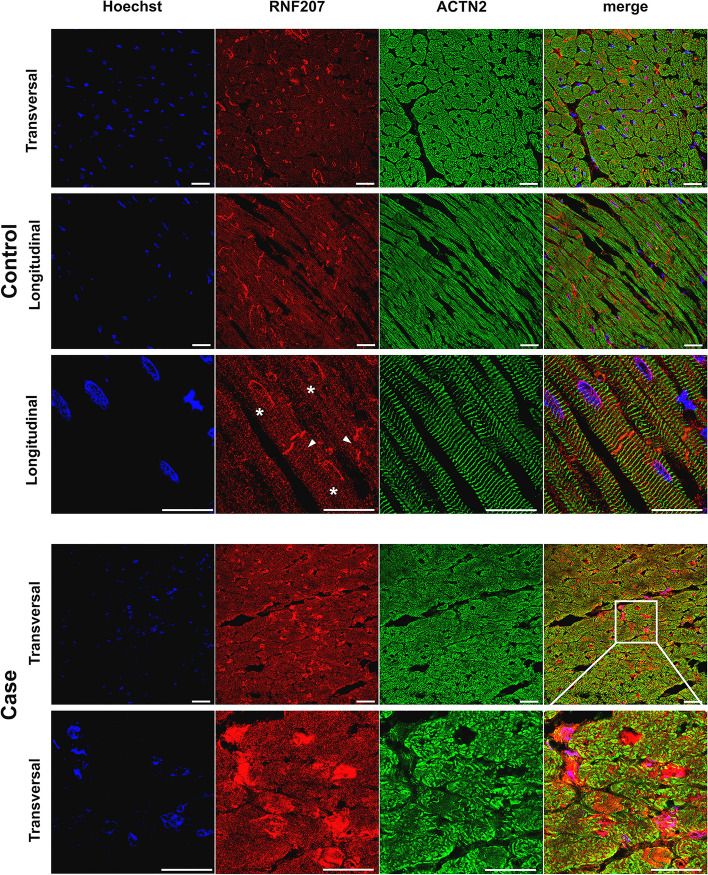
Results: Our results revealed two independent additive loci associated with the typical DCM phenotype comprising left ventricular systolic dysfunction and dilatation. We highlight two novel candidate genes, RNF207 and PRKAA2, known for their involvement in cardiac action potentials, energy homeostasis, and morphology. We further illustrate the distinct genetic etiologies underlying the typical DCM phenotype and ventricular premature contractions. Finally, we followed up on the canine discoveries in human DCM patients and discovered candidate variants in our two novel genes.
Conclusions: Collectively, our study yields insight into the molecular pathophysiology of DCM and provides a large animal model for preclinical studies.
Keywords: Arrhythmia; Cardiac; Cardiology; Companion animal; Complex trait; GWAS; Genetics; Transcriptomics.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
HL has consulted, and JD is an employee of Wisdom Panel, Kinship, which offers canine DNA testing as a commercial service. The remaining authors declare that they have no competing interests.
Figures