

Sympatho-adrenergic mechanisms in heart failure: new insights into pathophysiology
- PMID: 37724075
- PMCID: PMC10388789
- DOI: 10.1515/mr-2021-0007
Sympatho-adrenergic mechanisms in heart failure: new insights into pathophysiology
Abstract
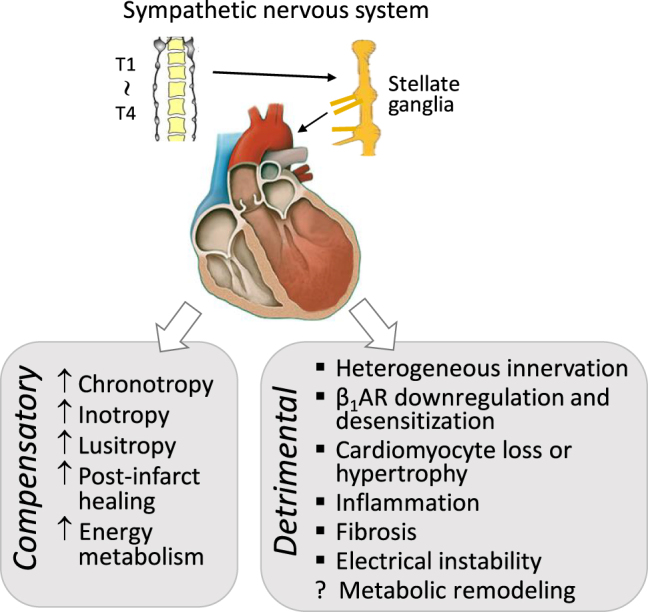
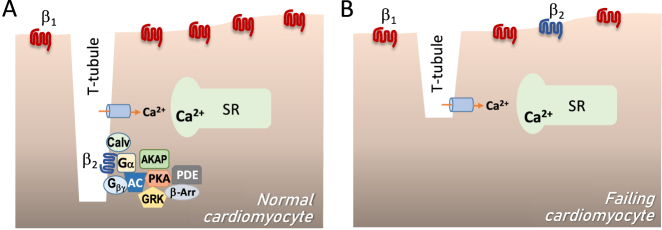
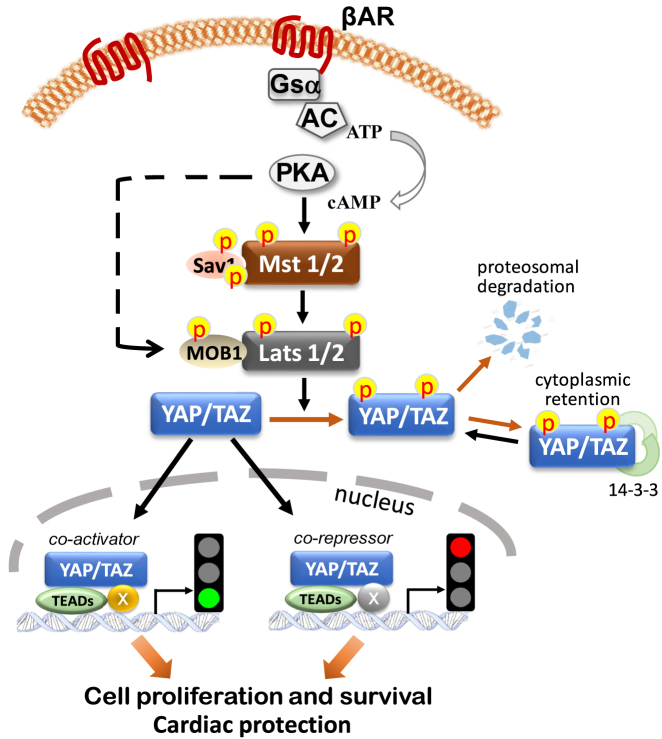
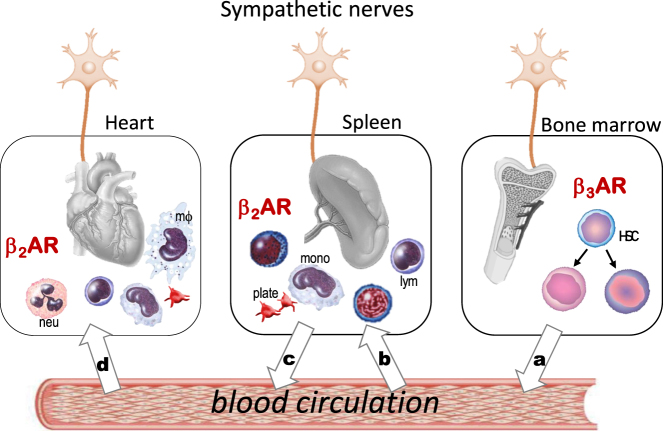
The sympathetic nervous system is activated in the setting of heart failure (HF) to compensate for hemodynamic instability. However, acute sympathetic surge or sustained high neuronal firing rates activates β-adrenergic receptor (βAR) signaling contributing to myocardial remodeling, dysfunction and electrical instability. Thus, sympatho-βAR activation is regarded as a hallmark of HF and forms pathophysiological basis for β-blocking therapy. Building upon earlier research findings, studies conducted in the recent decades have significantly advanced our understanding on the sympatho-adrenergic mechanism in HF, which forms the focus of this article. This review notes recent research progress regarding the roles of cardiac β2AR or α1AR in the failing heart, significance of β1AR-autoantibodies, and βAR signaling through G-protein independent signaling pathways. Sympatho-βAR regulation of immune cells or fibroblasts is specifically discussed. On the neuronal aspects, knowledge is assembled on the remodeling of sympathetic nerves of the failing heart, regulation by presynaptic α2AR of NE release, and findings on device-based neuromodulation of the sympathetic nervous system. The review ends with highlighting areas where significant knowledge gaps exist but hold promise for new breakthroughs.
Keywords: catecholamines; heart failure; sympathetic nervous system; β-adrenergic receptor; β-blocking therapy.
© 2021 Xiaojun Du, published by De Gruyter, Berlin/Boston.
Conflict of interest statement
Competing interests: The author has no conflict of interest to declare.
Figures
Similar articles
-
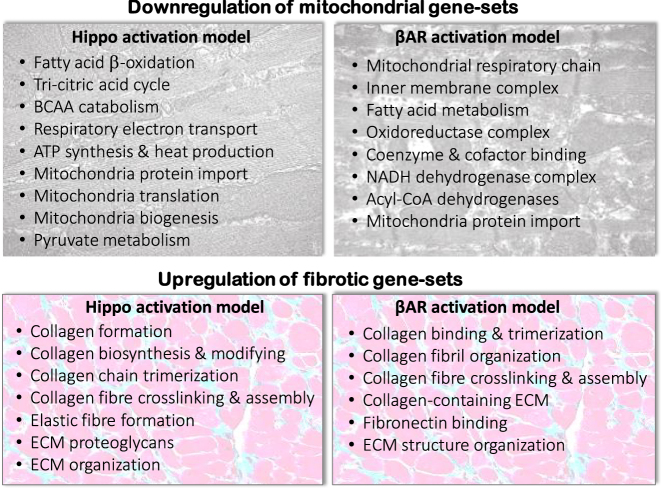
Coupling of β-adrenergic and Hippo pathway signaling: Implications for heart failure pathophysiology and metabolic therapy.Mitochondrion. 2024 Sep;78:101941. doi: 10.1016/j.mito.2024.101941. Epub 2024 Aug 7. Mitochondrion. 2024. PMID: 39122227 Review.
-
Therapeutic Targets for Treatment of Heart Failure: Focus on GRKs and β-Arrestins Affecting βAR Signaling.Front Pharmacol. 2018 Nov 27;9:1336. doi: 10.3389/fphar.2018.01336. eCollection 2018. Front Pharmacol. 2018. PMID: 30538631 Free PMC article. Review.
-
Sympatho-adrenergic activation of the ischemic myocardium and its arrhythmogenic impact.Herz. 1995 Jun;20(3):169-86. Herz. 1995. PMID: 7635399 Review.
-
Non-classical regulation of β1- and β 2-adrenoceptor-mediated inotropic responses in rat heart ventricle by the G protein Gi.Naunyn Schmiedebergs Arch Pharmacol. 2014 Dec;387(12):1177-86. doi: 10.1007/s00210-014-1036-7. Epub 2014 Sep 13. Naunyn Schmiedebergs Arch Pharmacol. 2014. PMID: 25216690
-
Gβγ-independent recruitment of G-protein coupled receptor kinase 2 drives tumor necrosis factor α-induced cardiac β-adrenergic receptor dysfunction.Circulation. 2013 Jul 23;128(4):377-87. doi: 10.1161/CIRCULATIONAHA.113.003183. Epub 2013 Jun 19. Circulation. 2013. PMID: 23785004 Free PMC article.
Cited by
-
Cardiomyopathy characterizing and heart failure risk predicting by echocardiography and pathoanatomy in aged male mice.Physiol Rep. 2024 Oct;12(20):e70061. doi: 10.14814/phy2.70061. Physiol Rep. 2024. PMID: 39411804 Free PMC article.
-
Cell-cell crosstalk in the heart.Med Rev (2021). 2022 Jan 11;1(1):3-5. doi: 10.1515/mr-2021-0029. eCollection 2021 Oct. Med Rev (2021). 2022. PMID: 37724073 Free PMC article. No abstract available.
-
Protective Effect of Uridine on Structural and Functional Rearrangements in Heart Mitochondria after a High-Dose Isoprenaline Exposure Modelling Stress-Induced Cardiomyopathy in Rats.Int J Mol Sci. 2023 Dec 9;24(24):17300. doi: 10.3390/ijms242417300. Int J Mol Sci. 2023. PMID: 38139129 Free PMC article.
References
-
- Kaye DM, Lambert GW, Lefkovits J, Morris M, Jennings G, Esler MD. Neurochemical evidence of cardiac sympathetic activation and increased central nervous system norepinephrine turnover in severe congestive heart failure. J Am Coll Cardiol. 1994;23:570–8. doi: 10.1016/0735-1097(94)90738-2. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous