

Amphipathic Helical Peptide L37pA Protects against Lung Vascular Endothelial Dysfunction Caused by Truncated Oxidized Phospholipids via Antagonism with CD36 Receptor
- PMID: 37725486
- PMCID: PMC10768836
- DOI: 10.1165/rcmb.2023-0127OC
Amphipathic Helical Peptide L37pA Protects against Lung Vascular Endothelial Dysfunction Caused by Truncated Oxidized Phospholipids via Antagonism with CD36 Receptor
Abstract
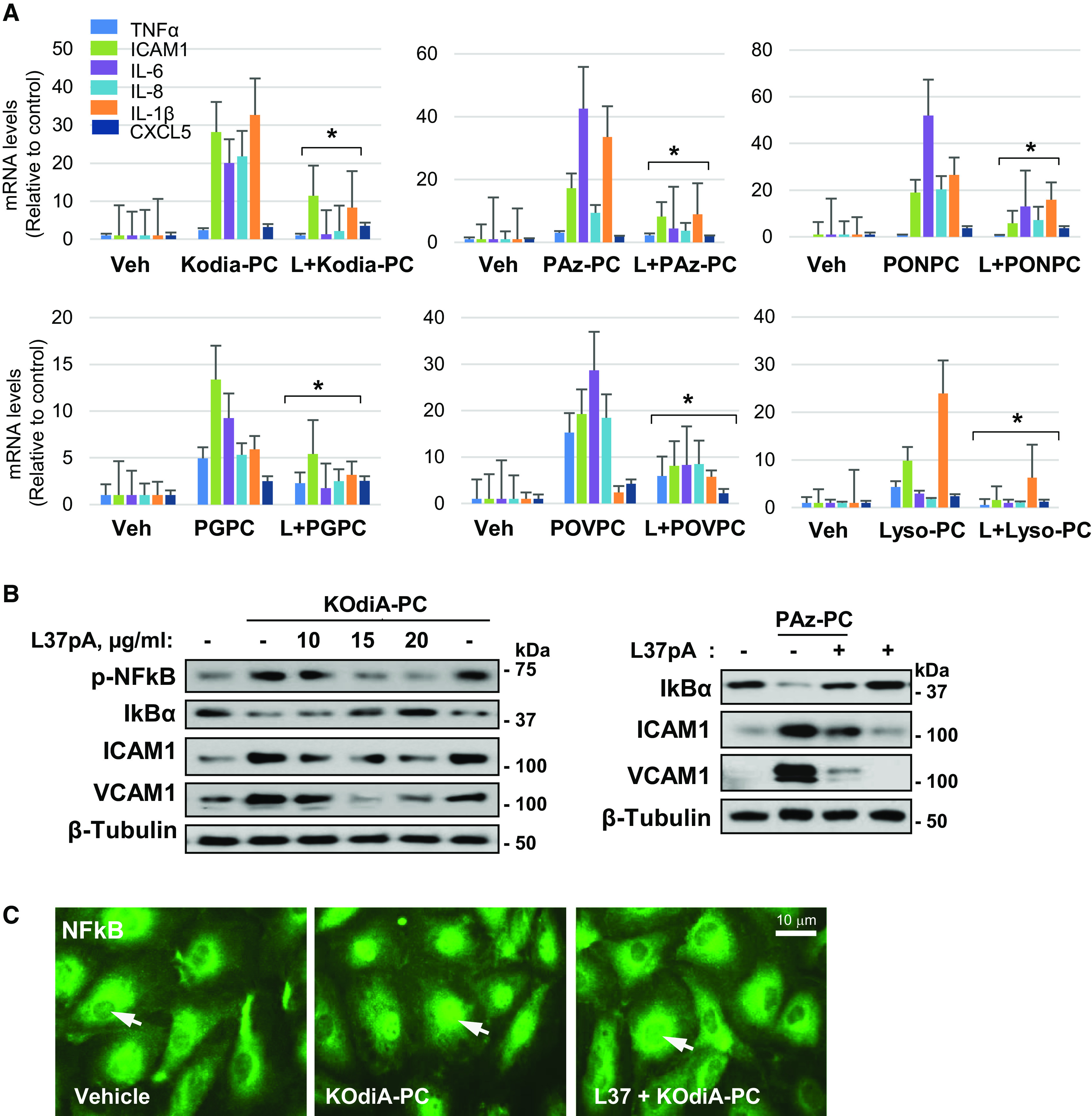
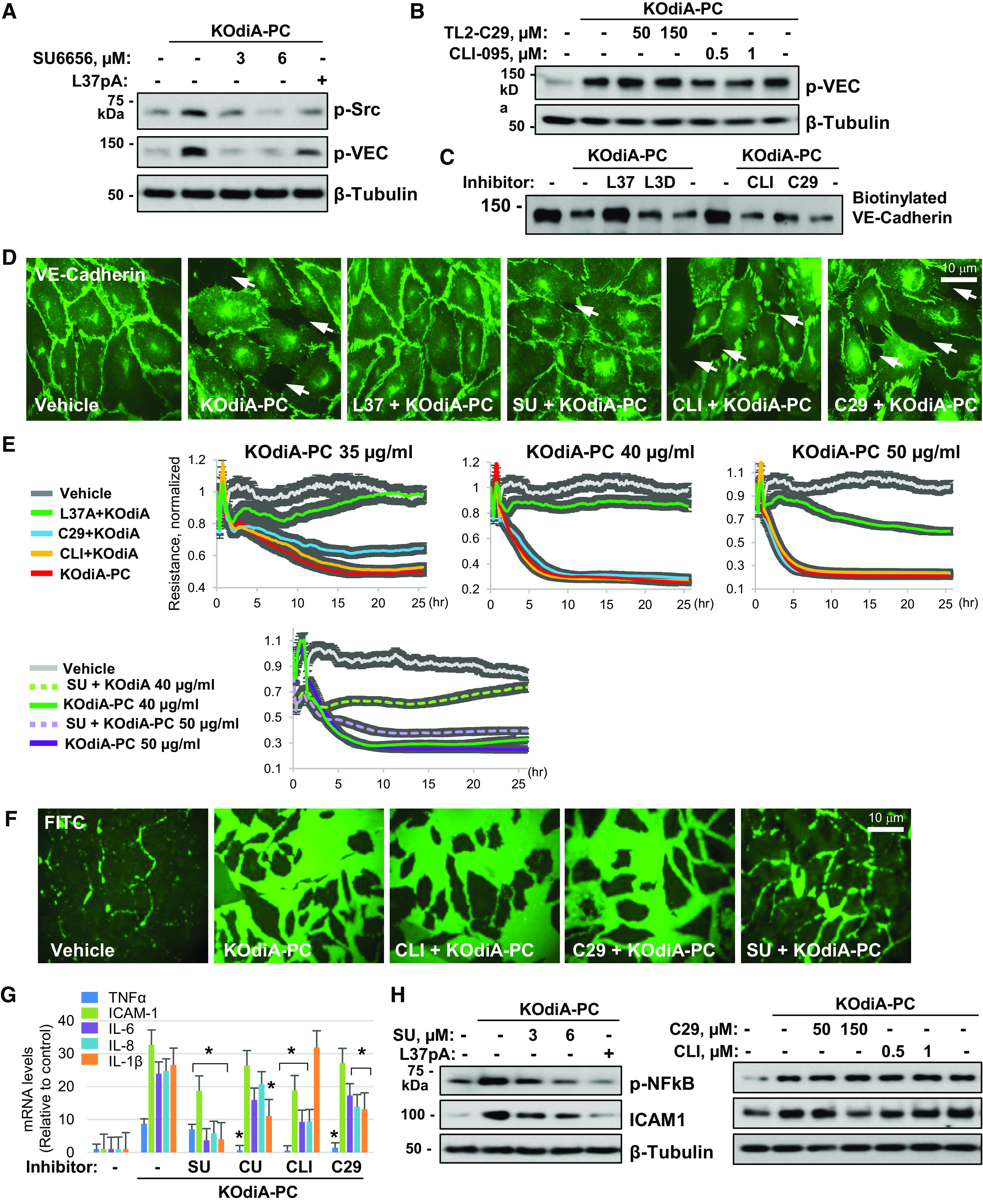
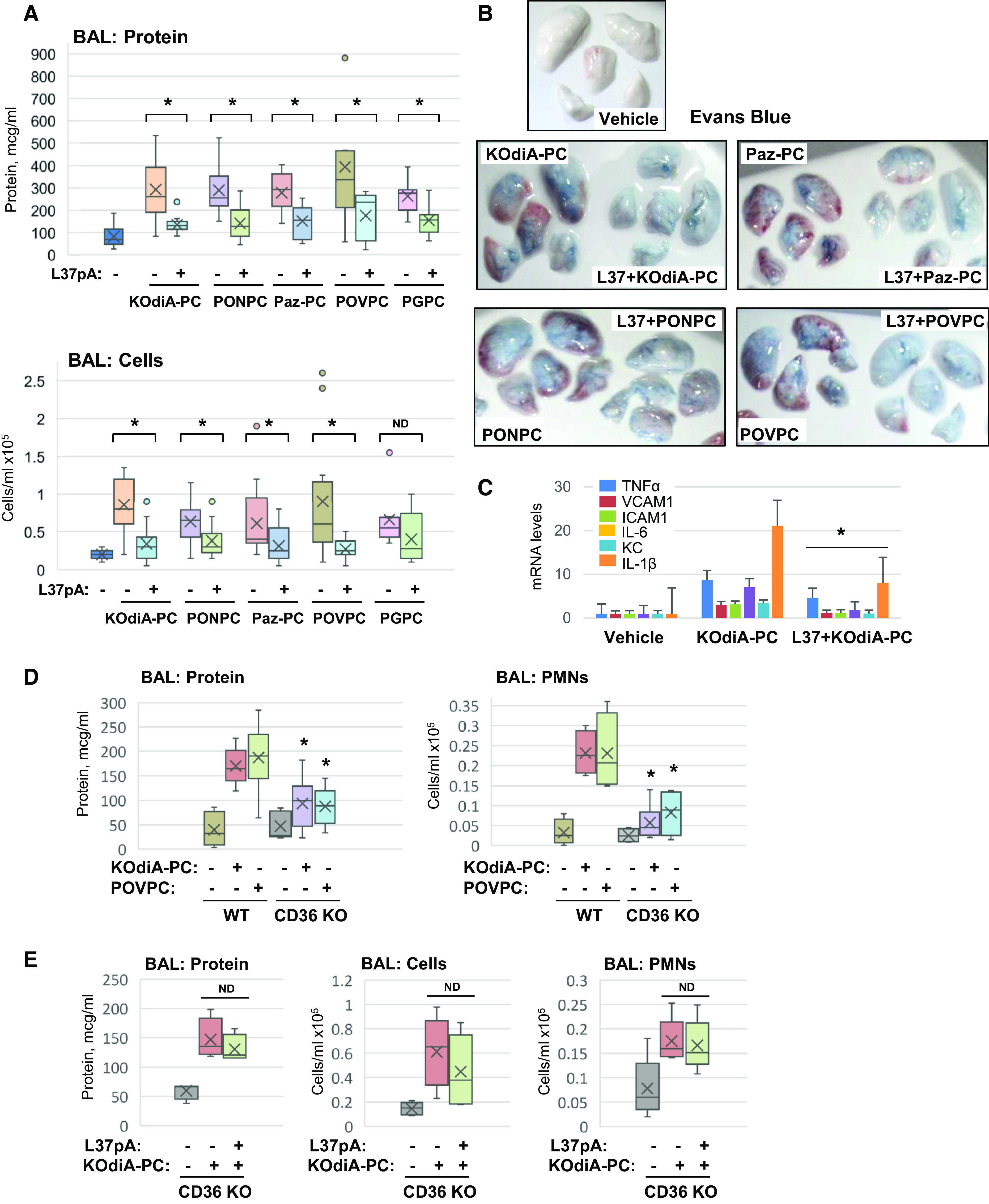
The generation of bioactive truncated oxidized phospholipids (Tr-OxPLs) from oxidation of cell-membrane or circulating lipoproteins is a common feature of various pathological states. Scavenger receptor CD36 is involved in lipid transport and acts as a receptor for Tr-OxPLs. Interestingly, Tr-OxPLs and CD36 are involved in endothelial dysfunction-derived acute lung injury, but the precise mechanistic connections remain unexplored. In the present study, we investigated the role of CD36 in mediating pulmonary endothelial cell (EC) dysfunction caused by Tr-OxPLs. Our results demonstrated that the Tr-OxPLs KOdia-PC, Paz-PC, PGPC, PON-PC, POV-PC, and lysophosphocholine caused an acute EC barrier disruption as revealed by measurements of transendothelial electrical resistance and VE-cadherin immunostaining. More importantly, a synthetic amphipathic helical peptide, L37pA, targeting human CD36 strongly attenuated Tr-OxPL-induced EC permeability. L37pA also suppressed Tr-OxPL-induced endothelial inflammatory activation monitored by mRNA expression of inflammatory cytokines/chemokines and adhesion molecules. In addition, L37pA blocked Tr-OxPL-induced NF-κB activation and tyrosine phosphorylation of Src kinase and VE-cadherin. The Src inhibitor SU6656 attenuated KOdia-PC-induced EC permeability and inflammation, but inhibition of the Toll-like receptors (TLRs) TLR1, TLR2, TLR4, and TLR6 had no such protective effects. CD36-knockout mice were more resistant to Tr-OxPL-induced lung injury. Treatment with L37pA was equally effective in ameliorating Tr-OxPL-induced vascular leak and lung inflammation as determined by an Evans blue extravasation assay and total cell and protein content in BAL fluid. Altogether, these results demonstrate an essential role of CD36 in mediating Tr-OxPL-induced EC dysfunction and suggest a strong therapeutic potential of CD36 inhibitory peptides in mitigating lung injury and inflammation.
Keywords: CD36; L37pA; acute lung injury; endothelial dysfunction; truncated oxidized phospholipids.
Figures




Comment in
-
Making Mountains out of Mole Hills: The Role of CD36 in Oxidized Phospholipid-driven Lung Injury.Am J Respir Cell Mol Biol. 2024 Jan;70(1):3-4. doi: 10.1165/rcmb.2023-0312ED. Am J Respir Cell Mol Biol. 2024. PMID: 37747355 Free PMC article. No abstract available.
References
-
- Bochkov V, Gesslbauer B, Mauerhofer C, Philippova M, Erne P, Oskolkova OV. Pleiotropic effects of oxidized phospholipids. Free Radic Biol Med . 2017;111:6–24. - PubMed
-
- Watson AD, Leitinger N, Navab M, Faull KF, Hörkkö S, Witztum JL, et al. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem . 1997;272:13597–13607. - PubMed
-
- Witztum JL, Berliner JA. Oxidized phospholipids and isoprostanes in atherosclerosis. Curr Opin Lipidol . 1998;9:441–448. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
