

The Next-Generation Oral Selective Estrogen Receptor Degrader Camizestrant (AZD9833) Suppresses ER+ Breast Cancer Growth and Overcomes Endocrine and CDK4/6 Inhibitor Resistance
- PMID: 37725704
- PMCID: PMC10690091
- DOI: 10.1158/0008-5472.CAN-23-0694
The Next-Generation Oral Selective Estrogen Receptor Degrader Camizestrant (AZD9833) Suppresses ER+ Breast Cancer Growth and Overcomes Endocrine and CDK4/6 Inhibitor Resistance
Abstract
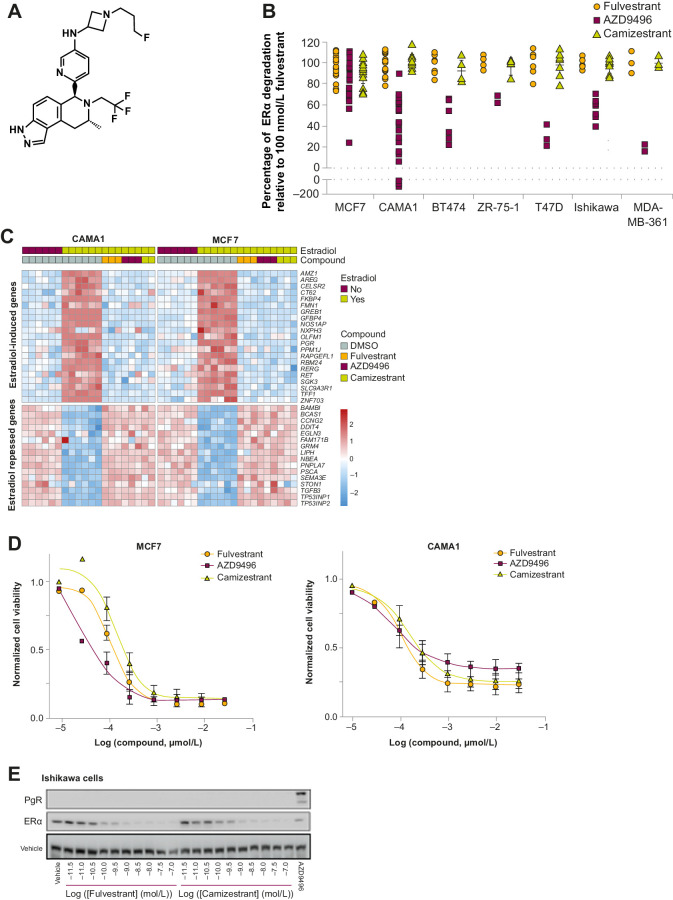
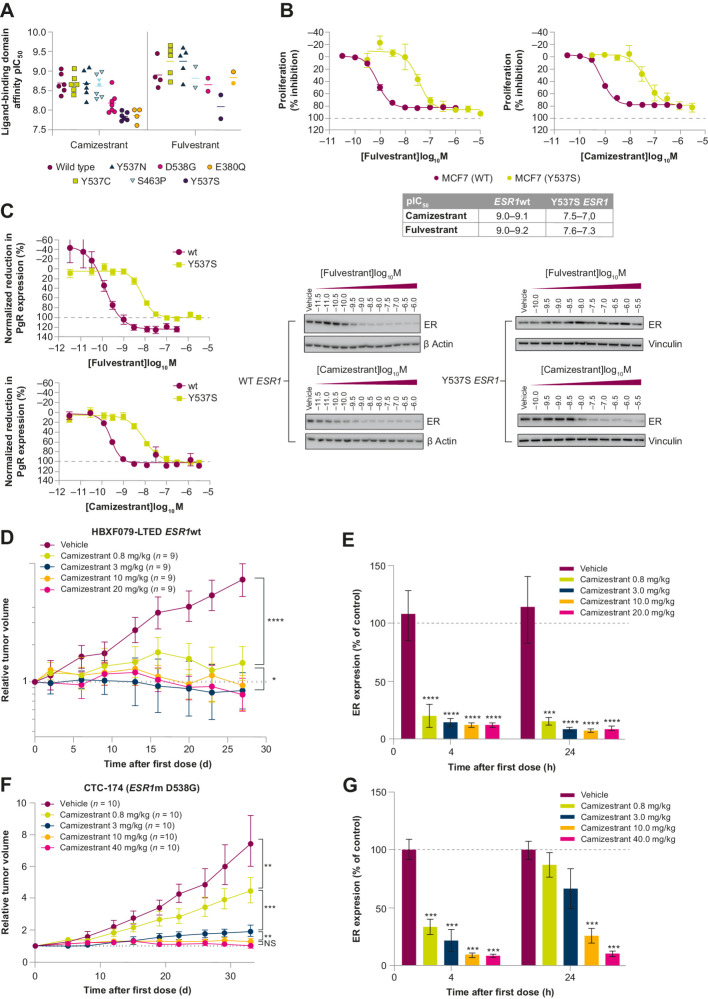
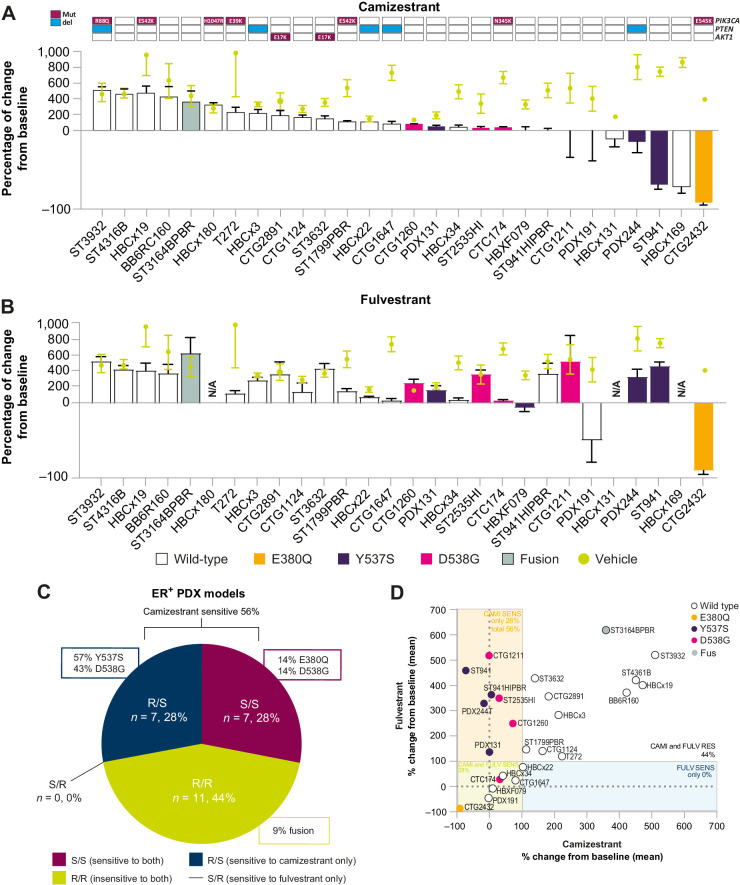
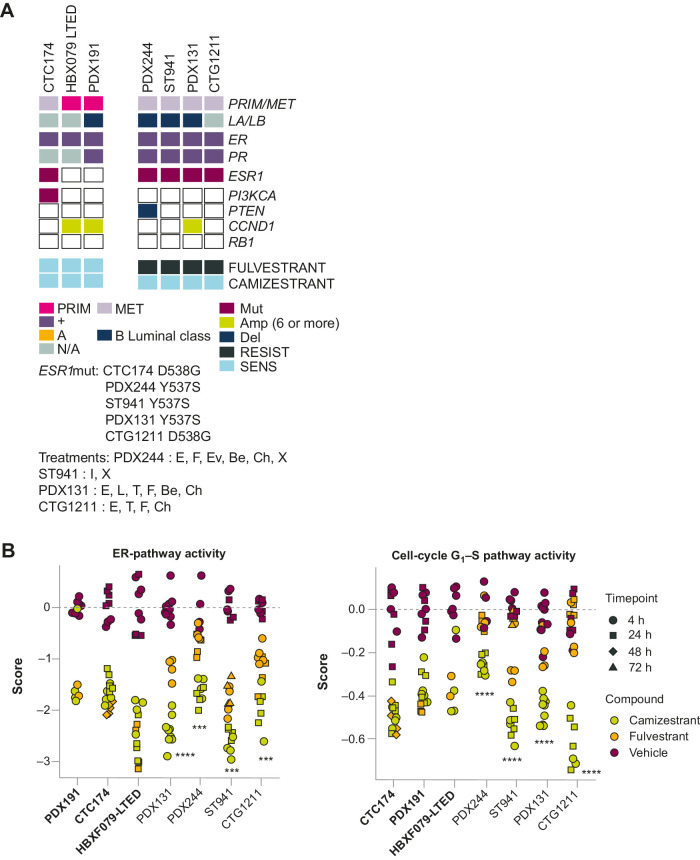
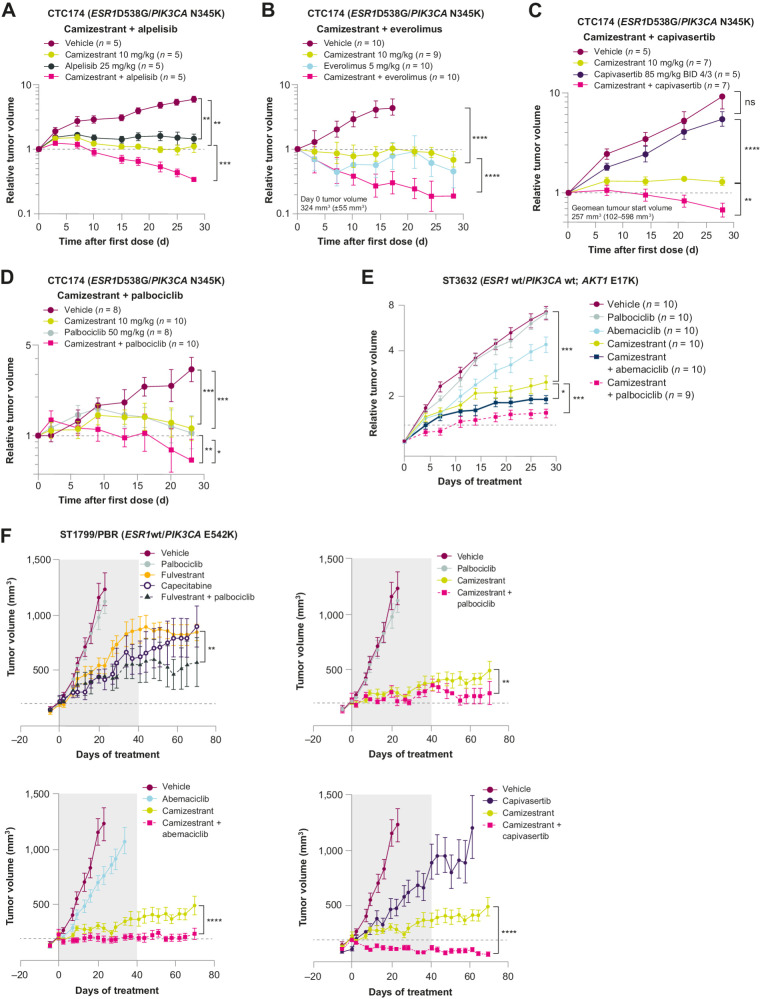
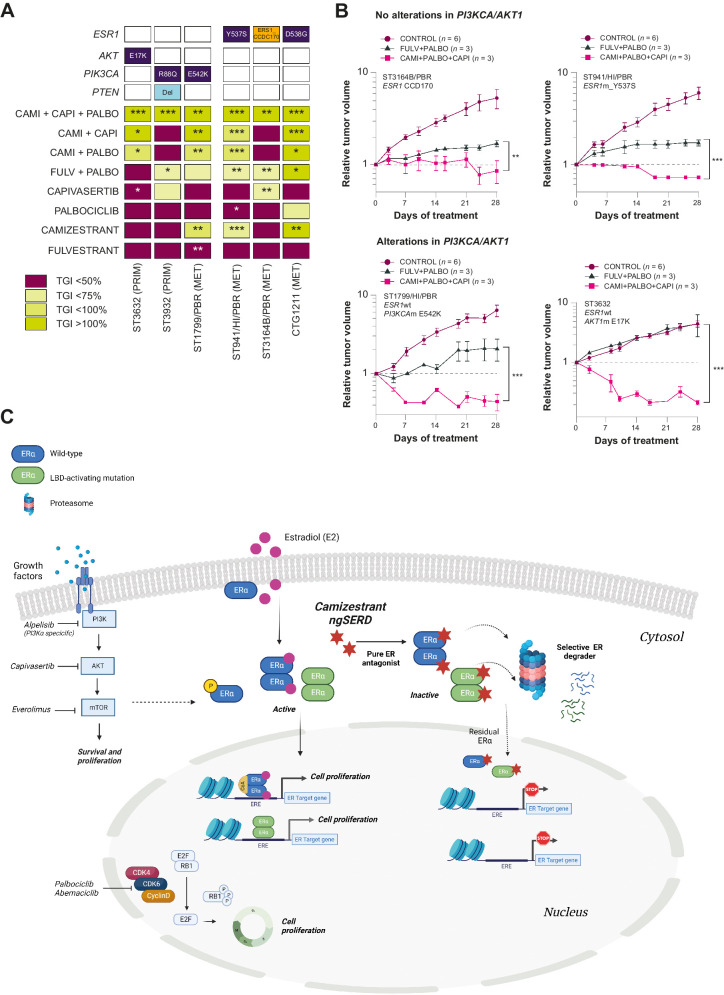
Oral selective estrogen receptor degraders (SERD) could become the backbone of endocrine therapy (ET) for estrogen receptor-positive (ER+) breast cancer, as they achieve greater inhibition of ER-driven cancers than current ETs and overcome key resistance mechanisms. In this study, we evaluated the preclinical pharmacology and efficacy of the next-generation oral SERD camizestrant (AZD9833) and assessed ER-co-targeting strategies by combining camizestrant with CDK4/6 inhibitors (CDK4/6i) and PI3K/AKT/mTOR-targeted therapy in models of progression on CDK4/6i and/or ET. Camizestrant demonstrated robust and selective ER degradation, modulated ER-regulated gene expression, and induced complete ER antagonism and significant antiproliferation activity in ESR1 wild-type (ESR1wt) and mutant (ESR1m) breast cancer cell lines and patient-derived xenograft (PDX) models. Camizestrant also delivered strong antitumor activity in fulvestrant-resistant ESR1wt and ESR1m PDX models. Evaluation of camizestrant in combination with CDK4/6i (palbociclib or abemaciclib) in CDK4/6-naive and -resistant models, as well as in combination with PI3Kαi (alpelisib), mTORi (everolimus), or AKTi (capivasertib), indicated that camizestrant was active with CDK4/6i or PI3K/AKT/mTORi and that antitumor activity was further increased by the triple combination. The response was observed independently of PI3K pathway mutation status. Overall, camizestrant shows strong and broad antitumor activity in ER+ breast cancer as a monotherapy and when combined with CDK4/6i and PI3K/AKT/mTORi.
Significance: Camizestrant, a next-generation oral SERD, shows promise in preclinical models of ER+ breast cancer alone and in combination with CDK4/6 and PI3K/AKT/mTOR inhibitors to address endocrine resistance, a current barrier to treatment.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Robertson JF, Blamey RW. The use of gonadotrophin-releasing hormone (GnRH) agonists in early and advanced breast cancer in pre- and perimenopausal women. Eur J Cancer 2003;39:861–9. - PubMed
-
- National Cancer Institute. 2020 01/12/2020. Drugs approved for breast cancer. National Cancer Institute. Available from:https://www.cancer.gov/about-cancer/treatment/drugs/breast. Accessed 2020 01/12/2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
