

Low glucose metabolite 3-phosphoglycerate switches PHGDH from serine synthesis to p53 activation to control cell fate
- PMID: 37726403
- PMCID: PMC10624847
- DOI: 10.1038/s41422-023-00874-4
Low glucose metabolite 3-phosphoglycerate switches PHGDH from serine synthesis to p53 activation to control cell fate
Abstract
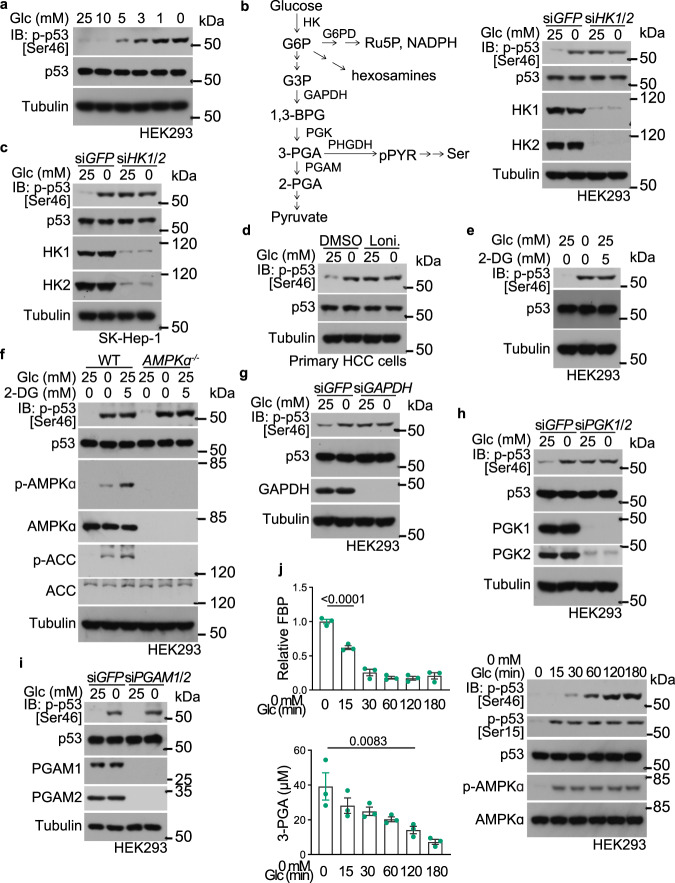
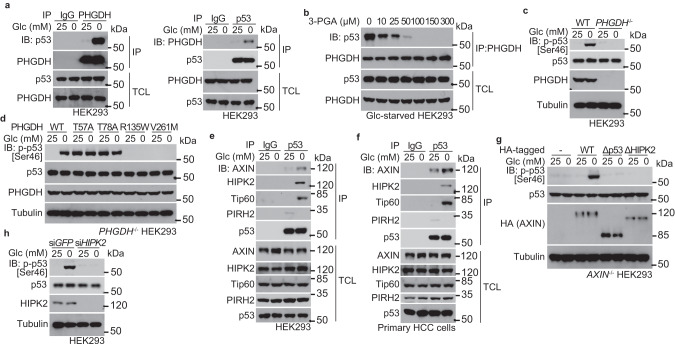
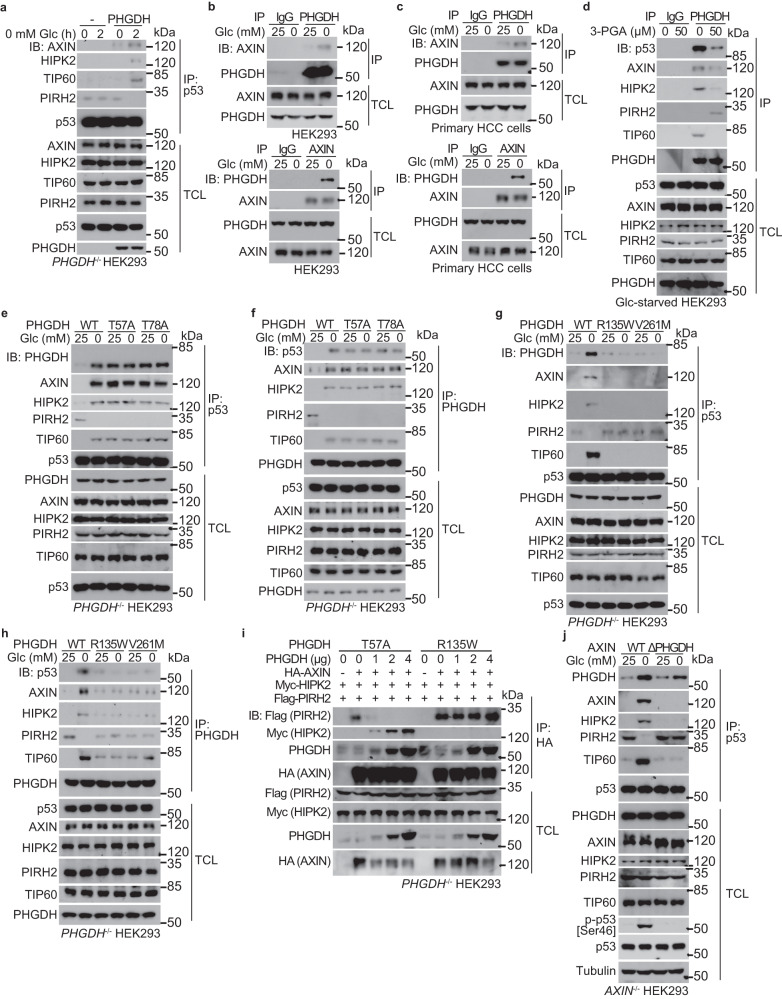
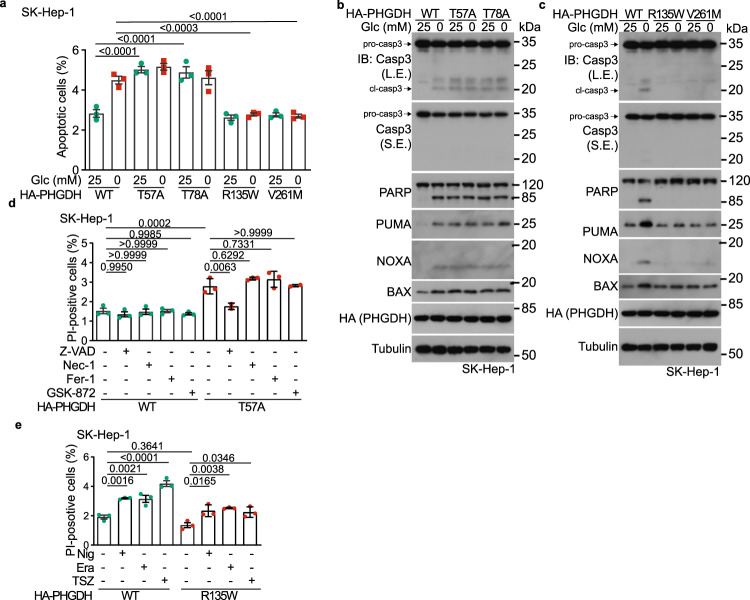
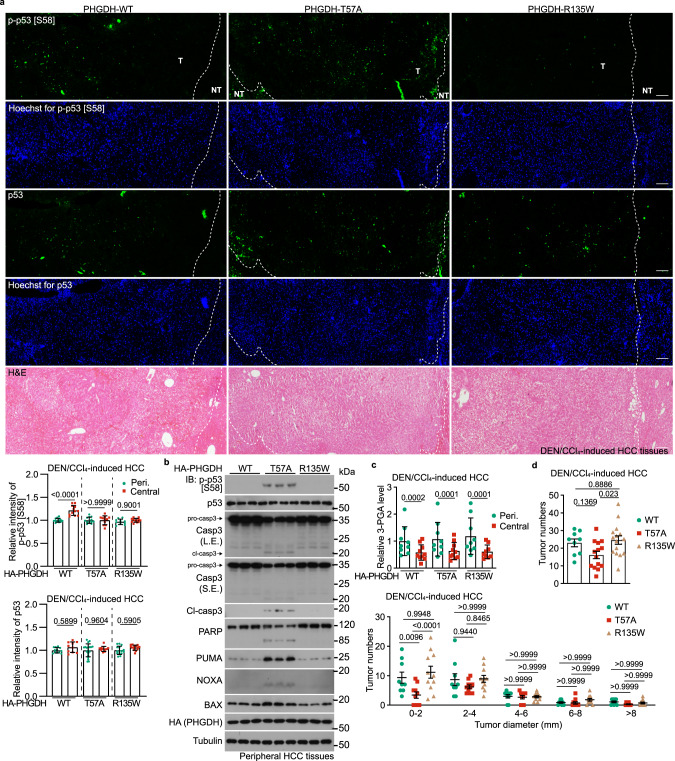
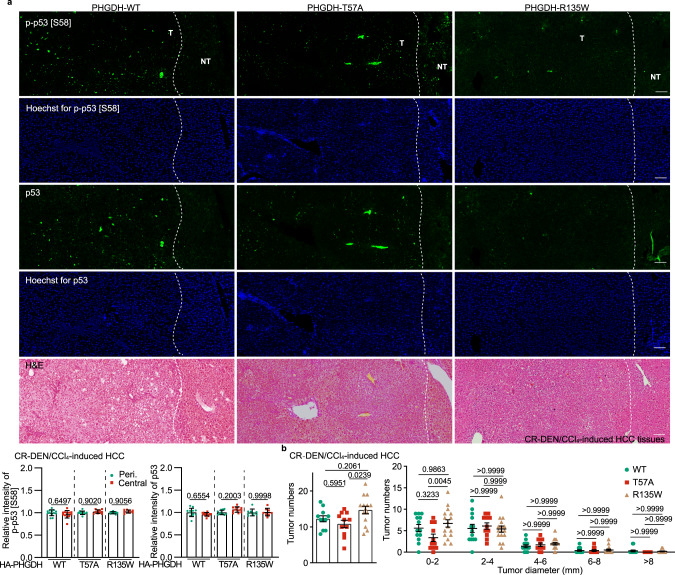
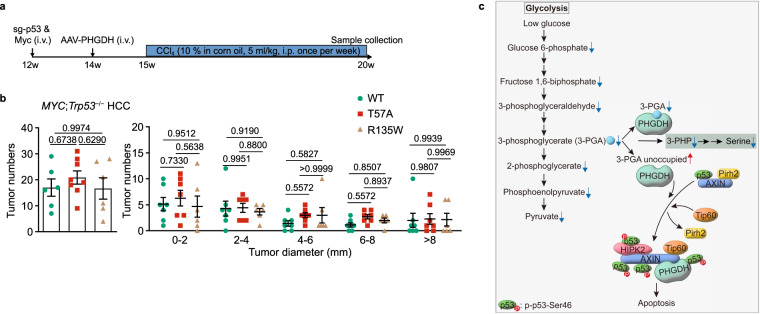
Glycolytic intermediary metabolites such as fructose-1,6-bisphosphate can serve as signals, controlling metabolic states beyond energy metabolism. However, whether glycolytic metabolites also play a role in controlling cell fate remains unexplored. Here, we find that low levels of glycolytic metabolite 3-phosphoglycerate (3-PGA) can switch phosphoglycerate dehydrogenase (PHGDH) from cataplerosis serine synthesis to pro-apoptotic activation of p53. PHGDH is a p53-binding protein, and when unoccupied by 3-PGA interacts with the scaffold protein AXIN in complex with the kinase HIPK2, both of which are also p53-binding proteins. This leads to the formation of a multivalent p53-binding complex that allows HIPK2 to specifically phosphorylate p53-Ser46 and thereby promote apoptosis. Furthermore, we show that PHGDH mutants (R135W and V261M) that are constitutively bound to 3-PGA abolish p53 activation even under low glucose conditions, while the mutants (T57A and T78A) unable to bind 3-PGA cause constitutive p53 activation and apoptosis in hepatocellular carcinoma (HCC) cells, even in the presence of high glucose. In vivo, PHGDH-T57A induces apoptosis and inhibits the growth of diethylnitrosamine-induced mouse HCC, whereas PHGDH-R135W prevents apoptosis and promotes HCC growth, and knockout of Trp53 abolishes these effects above. Importantly, caloric restriction that lowers whole-body glucose levels can impede HCC growth dependent on PHGDH. Together, these results unveil a mechanism by which glucose availability autonomously controls p53 activity, providing a new paradigm of cell fate control by metabolic substrate availability.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
