

[Congenital long QT syndrome]
- PMID: 37727265
- PMCID: PMC10506569
- DOI: 10.47487/apcyccv.v2i1.125
[Congenital long QT syndrome]
Abstract
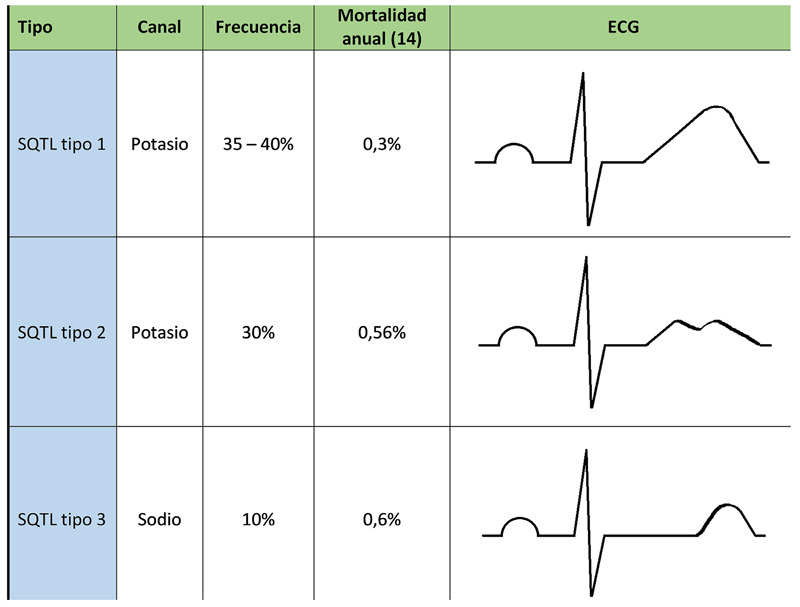
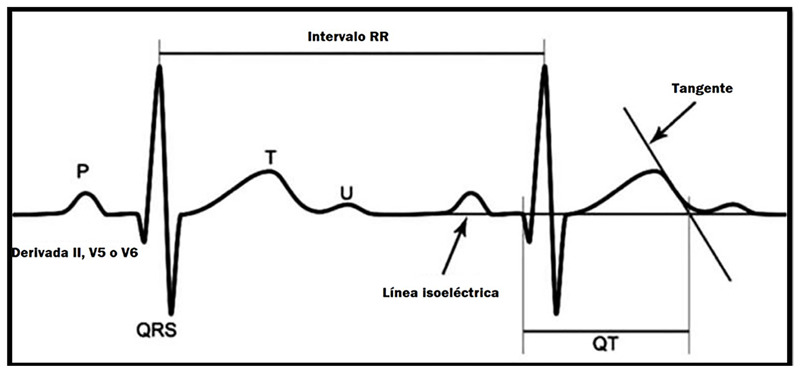
Congenital long QT syndrome (LQTS) represents a group of heart diseases of genetic origin characterized by prolongation of the QT interval and an abnormal T wave on the electrocardiogram (ECG). They can have a dominant or recessive expression, the latter associated with sensorineural deafness. In both cases, its clinical presentation is associated with recurrent syncope and sudden death as a consequence of ventricular tachycardia, specifically Torsades de Pointes. Currently they are classified according to the specific genetic defect, being able to compromise around 16 genes and almost 2000 mutations. It should be suspected in individuals with related symptoms, electrocardiographic findings, and family history. Management is based on the reduction or elimination of symptoms, and concomitantly the prevention of sudden death (SD), in those children with congenital deafness, the management requires the application of the otolaryngologist specialist's own measures. The cardiovascular management implies the modification of lifestyles, mainly the prohibition of competitive sports, including swimming, avoiding exposure to loud sounds or triggers. The medications used include beta-blockers, and more rarely flecainide, ranozaline, and verapamil; invasive management consists of the implantation of a cardioverter defibrillator or even left sympathetic denervation, each with its own risks and benefits. In any of the cases, we must avoid the circumstances that increase the QT interval, as well as carry out the appropriate analysis of the benefits and risks of each possible invasive measure.
El síndrome QT largo (SQTL) congénito representa un grupo de enfermedades cardiacas de origen genético, caracterizado por la prolongación del intervalo QT y una onda T anormal en el electrocardiograma (ECG). Pueden tener una expresión dominante o recesiva, esta última asociada con sordera neurosensorial. En ambos casos su presentación clínica está asociada con síncopes recurrentes y muerte súbita como consecuencia de una taquicardia ventricular, específicamente a torsades de pointes. Actualmente se clasifican en función del defecto genético específico, pudiendo comprometer alrededor de 16 genes y casi 2000 mutaciones. Debe ser sospechada en individuos con la clínica relacionada, hallazgos electrocardiográficos y antecedentes familiares. El manejo está basado en la disminución o eliminación de los síntomas y, concomitantemente, la prevención de la muerte súbita (MS), en aquellos niños con sordera congénita el manejo requiere la aplicación de medidas propias del otorrinolaringólogo. Desde el punto de vista cardiovascular, el manejo implica la modificación de estilos de vida, principalmente la prohibición de deportes competitivos, entre ellos la natación, evitar la exposición a sonidos intensos o factores desencadenantes. La medicación usada abarca a los betabloqueadores y, más raramente, flecainida, ranozalina y verapamilo; el manejo invasivo consiste el implante de un cardiodesfibrilador o, incluso, la denervación simpática izquierda, cada una de ellas con sus propios riesgos y beneficios. En cualquiera de los casos debemos evitar las circunstancias que incrementen el intervalo QT, así como realizar el adecuado análisis de los beneficios y riesgos de cada posible medida invasiva.
Keywords: Death, Sudden; Long QT Syndrome; Pediatrics; Torsades de Pointes.
Conflict of interest statement
Conflictos de interés: Los autores declaran no tener ningún conflicto de interés.
Figures
Similar articles
-
Evaluation and Management of Athletes With Long QT Syndrome.Sports Health. 2016 Nov/Dec;8(6):527-535. doi: 10.1177/1941738116660294. Epub 2016 Aug 6. Sports Health. 2016. PMID: 27480102 Free PMC article. Review.
-
Management of Patients with Long QT Syndrome.Korean Circ J. 2016 Nov;46(6):747-752. doi: 10.4070/kcj.2016.46.6.747. Epub 2016 Oct 20. Korean Circ J. 2016. PMID: 27826330 Free PMC article. Review.
-
Long QT syndrome and short QT syndrome.Prog Cardiovasc Dis. 2008 Nov-Dec;51(3):264-78. doi: 10.1016/j.pcad.2008.10.006. Prog Cardiovasc Dis. 2008. PMID: 19026859 Review.
-
Mechanisms and clinical management of inherited channelopathies: long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome.Heart Rhythm. 2009 Aug;6(8 Suppl):S51-5. doi: 10.1016/j.hrthm.2009.02.009. Epub 2009 Feb 12. Heart Rhythm. 2009. PMID: 19631908 Review.
-
Congenital and acquired long QT syndrome. Current concepts and management.Cardiol Rev. 2004 Jul-Aug;12(4):222-34. doi: 10.1097/01.crd.0000123842.42287.cf. Cardiol Rev. 2004. PMID: 15191637 Review.
References
-
- Brugada J, Blom N, Sarquella-Brugada G, Blomstrom-Lundqvist C, Deanfield J, Janousek J, et al. Pharmacological and non-pharmacological therapy for arrhythmias in the pediatric population EHRA and AEPC-Arrhythmia Working Group joint consensus statement. EP Eur. 2013;15(9):1337–1382. doi: 10.1093/europace/eut082. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources