

Systematically testing human HMBS missense variants to reveal mechanism and pathogenic variation
- PMID: 37729906
- PMCID: PMC10577081
- DOI: 10.1016/j.ajhg.2023.08.012
Systematically testing human HMBS missense variants to reveal mechanism and pathogenic variation
Abstract
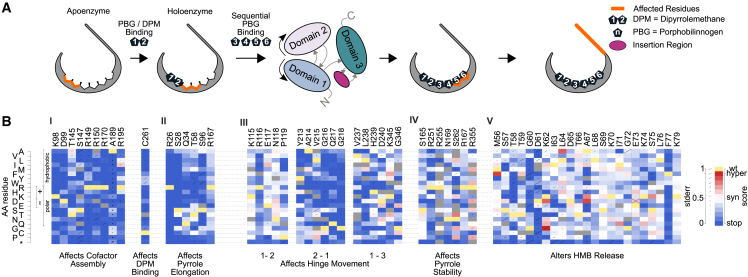
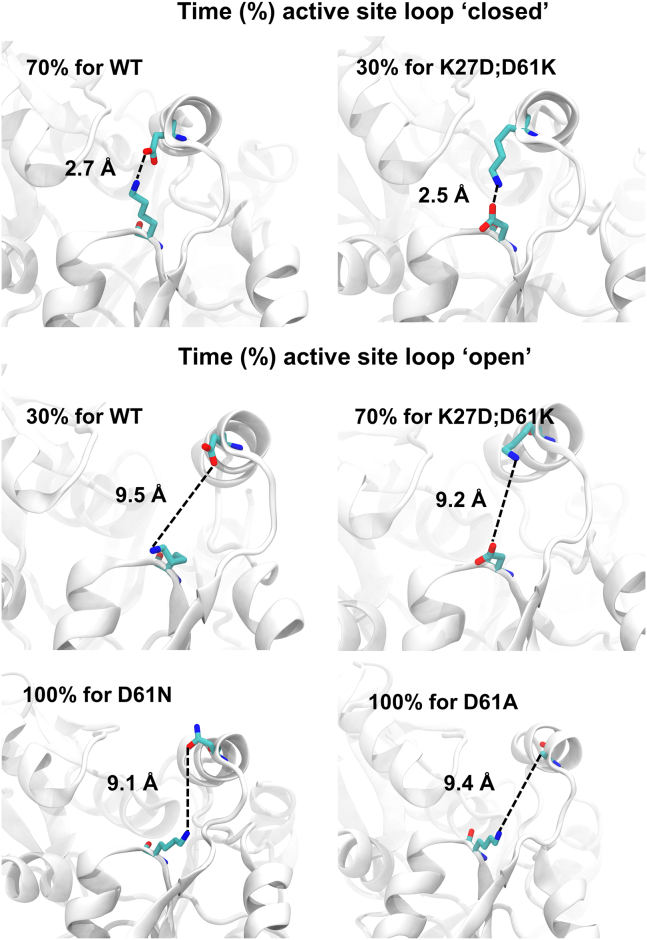
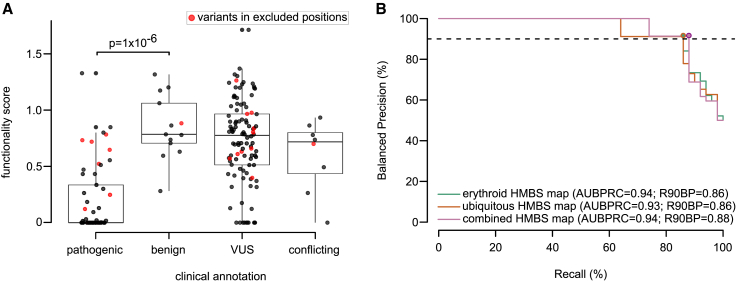
Defects in hydroxymethylbilane synthase (HMBS) can cause acute intermittent porphyria (AIP), an acute neurological disease. Although sequencing-based diagnosis can be definitive, ∼⅓ of clinical HMBS variants are missense variants, and most clinically reported HMBS missense variants are designated as "variants of uncertain significance" (VUSs). Using saturation mutagenesis, en masse selection, and sequencing, we applied a multiplexed validated assay to both the erythroid-specific and ubiquitous isoforms of HMBS, obtaining confident functional impact scores for >84% of all possible amino acid substitutions. The resulting variant effect maps generally agreed with biochemical expectations and provide further evidence that HMBS can function as a monomer. Additionally, the maps implicated specific residues as having roles in active site dynamics, which was further supported by molecular dynamics simulations. Most importantly, these maps can help discriminate pathogenic from benign HMBS variants, proactively providing evidence even for yet-to-be-observed clinical missense variants.
Keywords: AIP; HMBS,; acute hepatic porphryia; acute intermittent porphryia; clinical variant interpretation; deep mutational scanning; heme biosynthesis; hydroxymethylbilane synthase; molecular dynamics; variant effect mapping.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.P.R. is an investor in Ranomics, Inc., and is an investor in and advisor for SeqWell, Inc., BioSymetrics, Inc., and Constantiam Biosciences, Inc., and has accepted conference travel support from Illumina, Inc. L.F., A.C., and R.N. are employed by and invested in Invitae. R.J.D. has received both a grant and royalties and has also served as a consultant for Alnylam Pharmaceuticals.
Figures


Update of
-
Systematically testing human HMBS missense variants to reveal mechanism and pathogenic variation.bioRxiv [Preprint]. 2023 Feb 6:2023.02.06.527353. doi: 10.1101/2023.02.06.527353. bioRxiv. 2023. Update in: Am J Hum Genet. 2023 Oct 5;110(10):1769-1786. doi: 10.1016/j.ajhg.2023.08.012. PMID: 36798224 Free PMC article. Updated. Preprint.
References
-
- Bissell D.M., Anderson K.E., Bonkovsky H.L. N. Engl. J. Med. 2017;377:862–872. - PubMed
-
- Chen B., Solis-Villa C., Hakenberg J., Qiao W., Srinivasan R.R., Yasuda M., Balwani M., Doheny D., Peter I., Chen R., Desnick R.J. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum. Mutat. 2016;37:1215–1222. - PMC - PubMed
-
- Baumann K., Kauppinen R. Penetrance and predictive value of genetic screening in acute porphyria. Mol. Genet. Metab. 2020;130:87–99. - PubMed
-
- Lenglet H., Schmitt C., Grange T., Manceau H., Karboul N., Bouchet-Crivat F., Robreau A.-M., Nicolas G., Lamoril J., Simonin S., et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum. Mol. Genet. 2018;27:1164–1173. - PubMed
-
- Grandchamp B., De Verneuil H., Beaumont C., Chretien S., Walter O., Nordmann Y. Tissue-specific expression of porphobilinogen deaminase. Two isoenzymes from a single gene. Eur. J. Biochem. 1987;162:105–110. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
