

Stepwise emergence of the neuronal gene expression program in early animal evolution
- PMID: 37729907
- PMCID: PMC10580291
- DOI: 10.1016/j.cell.2023.08.027
Stepwise emergence of the neuronal gene expression program in early animal evolution
Abstract
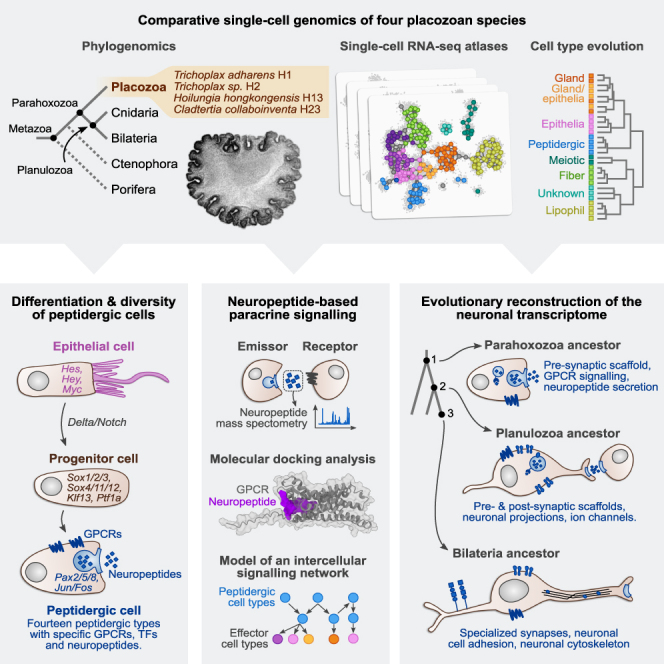
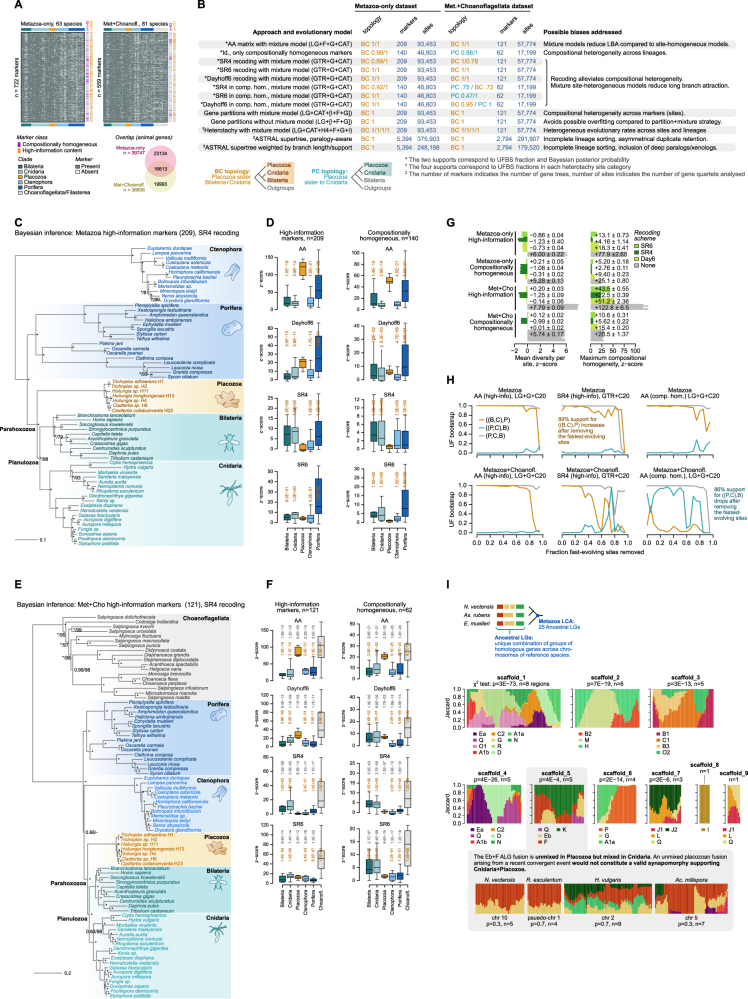
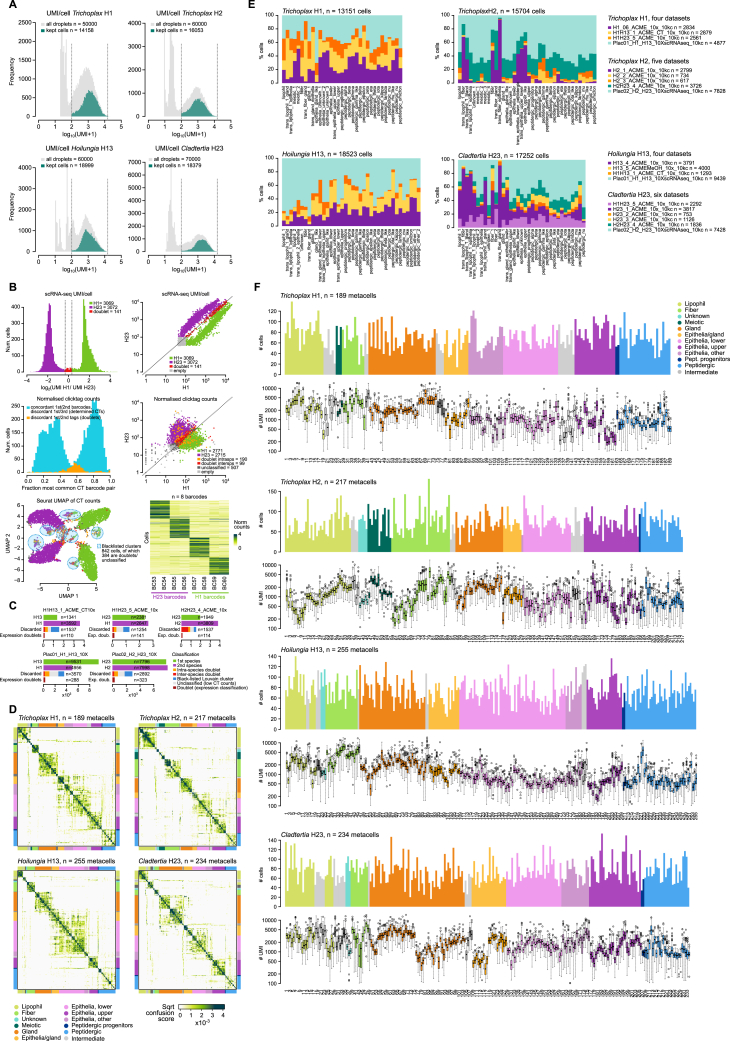
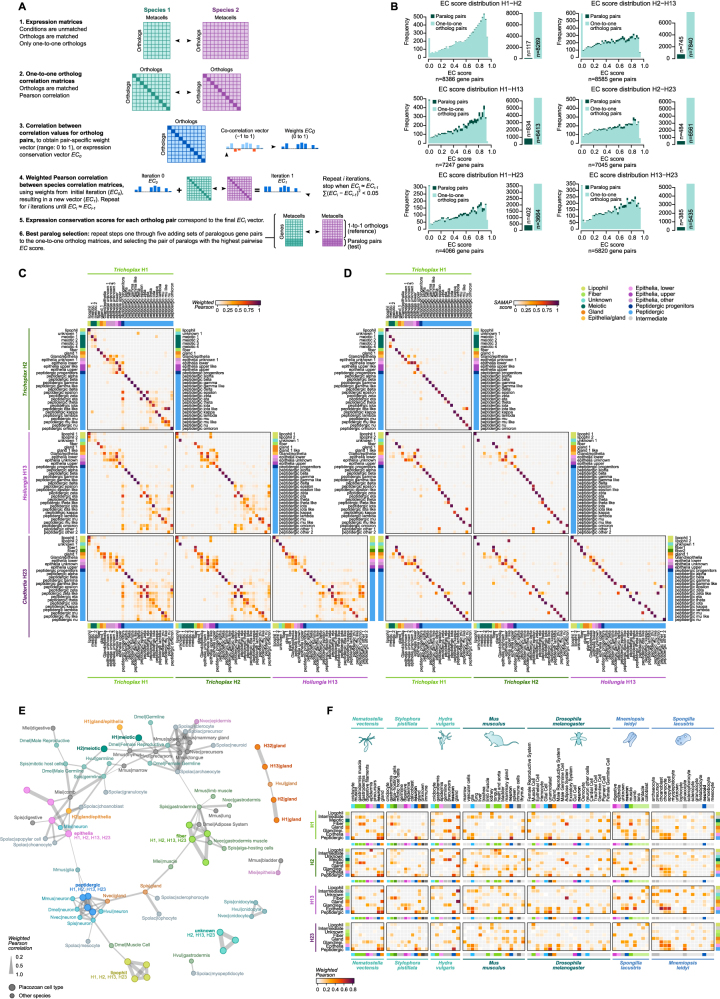
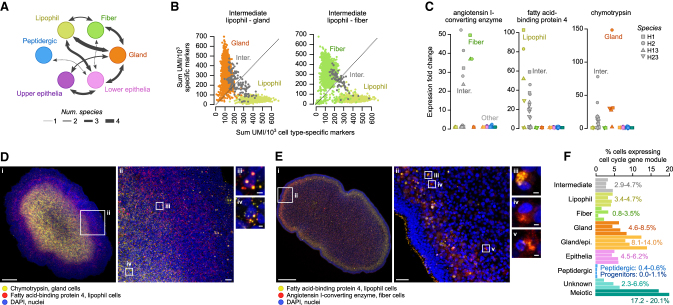
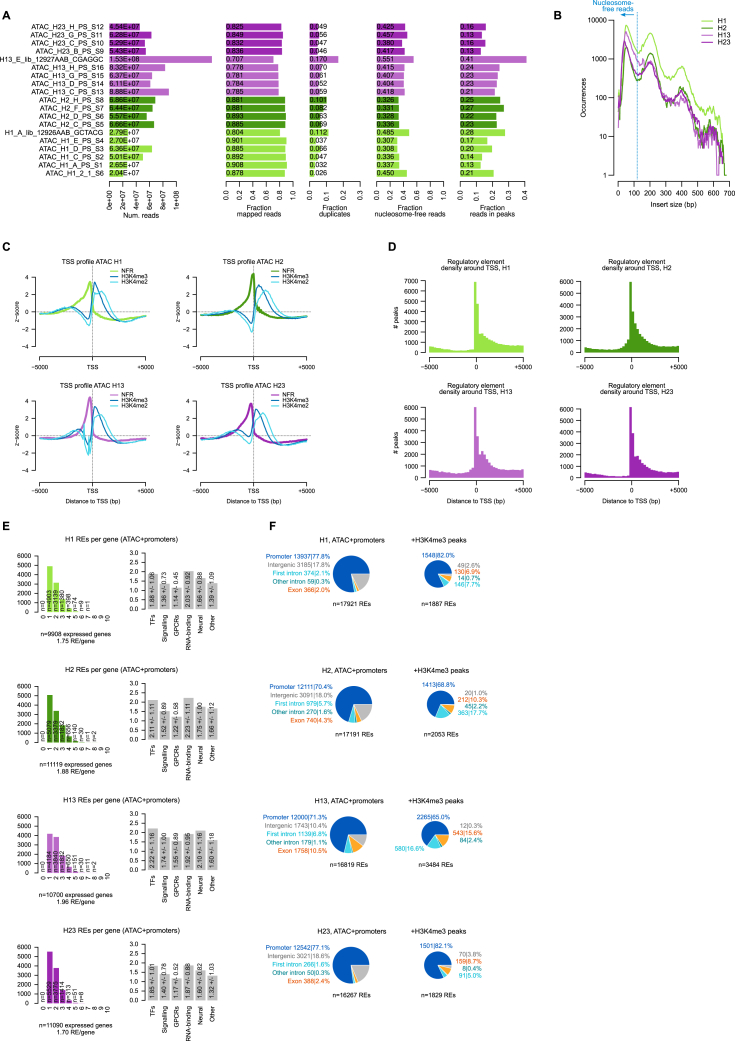
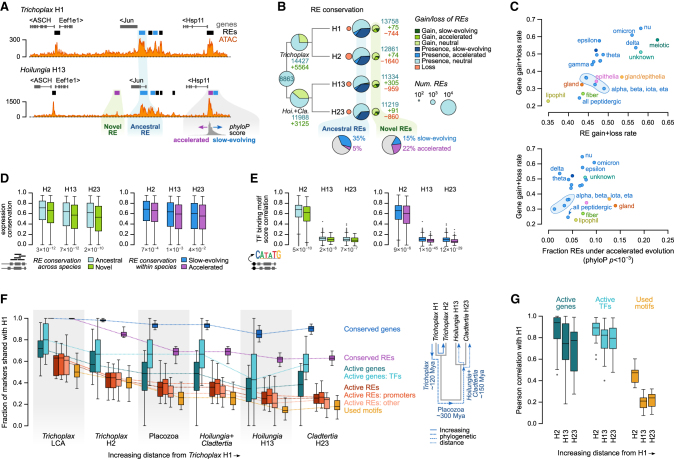
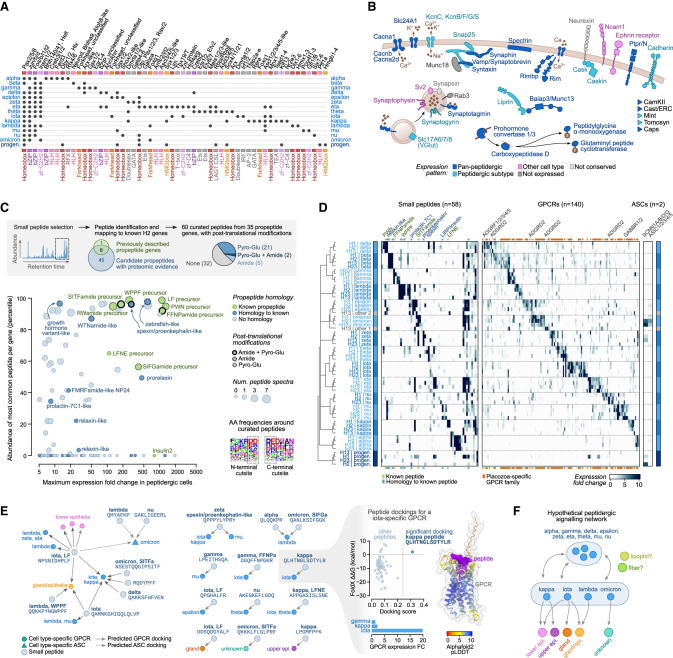
The assembly of the neuronal and other major cell type programs occurred early in animal evolution. We can reconstruct this process by studying non-bilaterians like placozoans. These small disc-shaped animals not only have nine morphologically described cell types and no neurons but also show coordinated behaviors triggered by peptide-secreting cells. We investigated possible neuronal affinities of these peptidergic cells using phylogenetics, chromatin profiling, and comparative single-cell genomics in four placozoans. We found conserved cell type expression programs across placozoans, including populations of transdifferentiating and cycling cells, suggestive of active cell type homeostasis. We also uncovered fourteen peptidergic cell types expressing neuronal-associated components like the pre-synaptic scaffold that derive from progenitor cells with neurogenesis signatures. In contrast, earlier-branching animals like sponges and ctenophores lacked this conserved expression. Our findings indicate that key neuronal developmental and effector gene modules evolved before the advent of cnidarian/bilaterian neurons in the context of paracrine cell signaling.
Keywords: Notch signaling; biodiversity; cell differentiation; chromatin biology; comparative genomics; developmental biology; evolution; neuroscience; phylogenetics; single-cell transcriptomics.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
