

This is a preprint.
ParSE-seq: A Calibrated Multiplexed Assay to Facilitate the Clinical Classification of Putative Splice-altering Variants
- PMID: 37732247
- PMCID: PMC10508793
- DOI: 10.1101/2023.09.04.23295019
ParSE-seq: A Calibrated Multiplexed Assay to Facilitate the Clinical Classification of Putative Splice-altering Variants
Update in
-
ParSE-seq: a calibrated multiplexed assay to facilitate the clinical classification of putative splice-altering variants.Nat Commun. 2024 Sep 27;15(1):8320. doi: 10.1038/s41467-024-52474-4. Nat Commun. 2024. PMID: 39333091 Free PMC article.
Abstract
Background: Interpreting the clinical significance of putative splice-altering variants outside 2-base pair canonical splice sites remains difficult without functional studies.
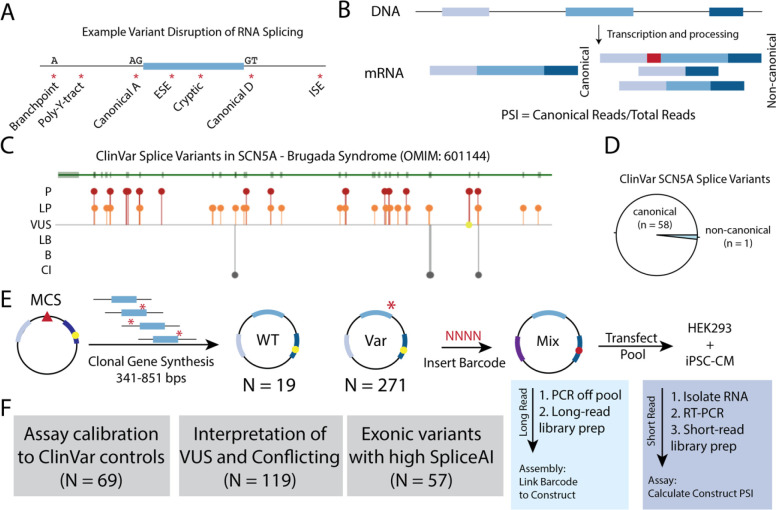
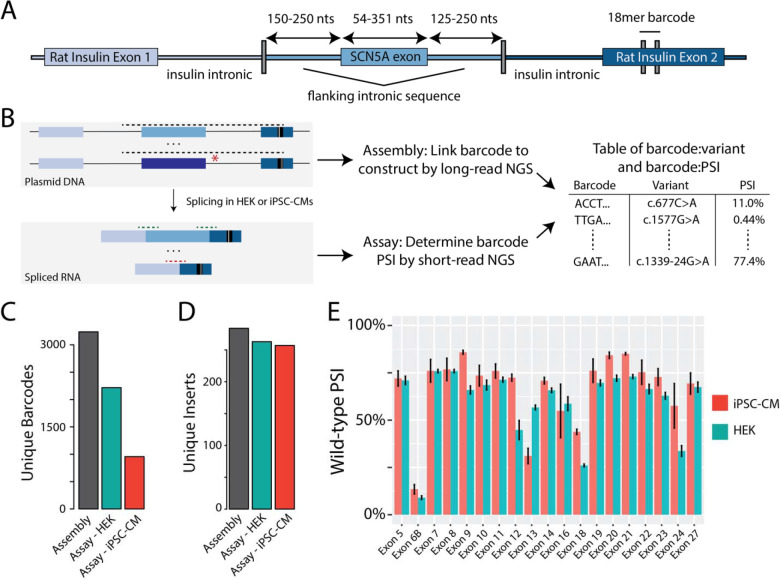
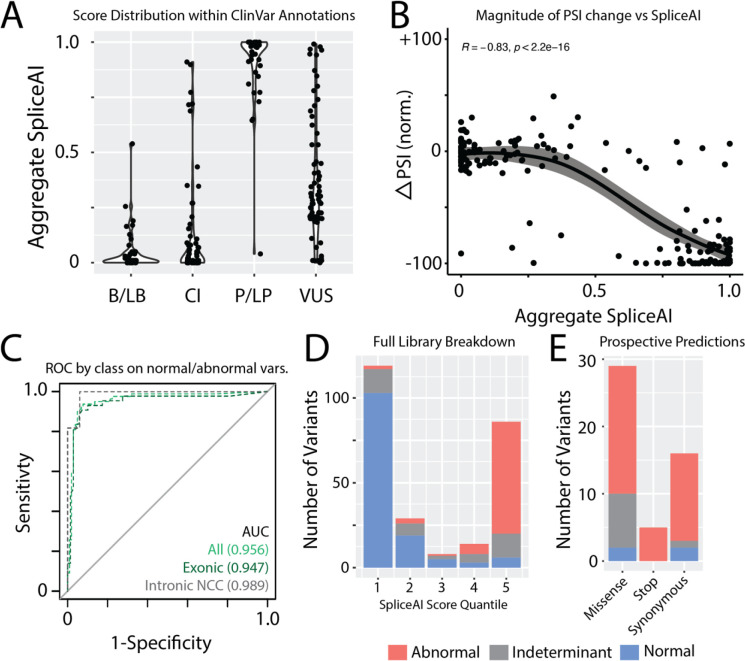
Methods: We developed Parallel Splice Effect Sequencing (ParSE-seq), a multiplexed minigene-based assay, to test variant effects on RNA splicing quantified by high-throughput sequencing. We studied variants in SCN5A, an arrhythmia-associated gene which encodes the major cardiac voltage-gated sodium channel. We used the computational tool SpliceAI to prioritize exonic and intronic candidate splice variants, and ClinVar to select benign and pathogenic control variants. We generated a pool of 284 barcoded minigene plasmids, transfected them into Human Embryonic Kidney (HEK293) cells and induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs), sequenced the resulting pools of splicing products, and calibrated the assay to the American College of Medical Genetics and Genomics scheme. Variants were interpreted using the calibrated functional data, and experimental data were compared to SpliceAI predictions. We further studied some splice-altering missense variants by cDNA-based automated patch clamping (APC) in HEK cells and assessed splicing and sodium channel function in CRISPR-edited iPSC-CMs.
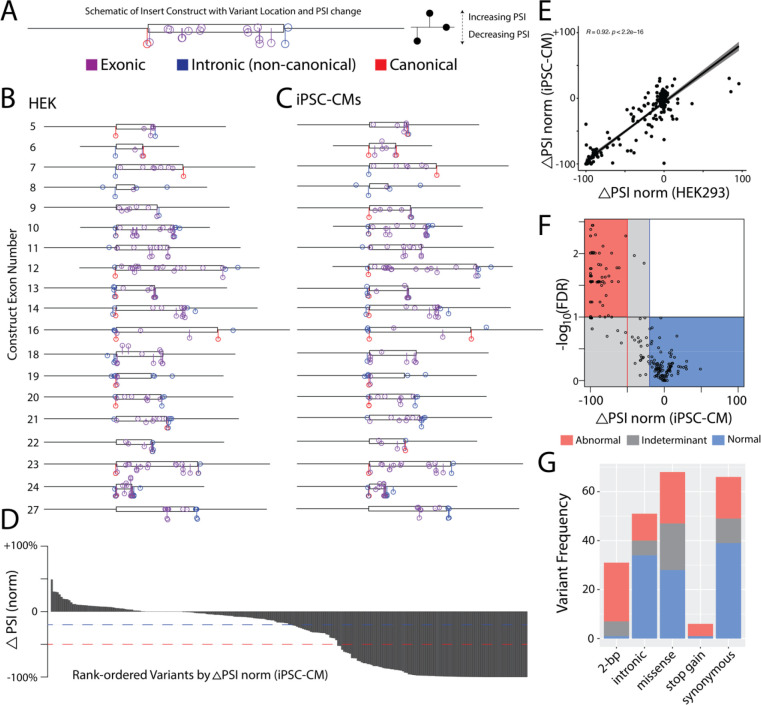
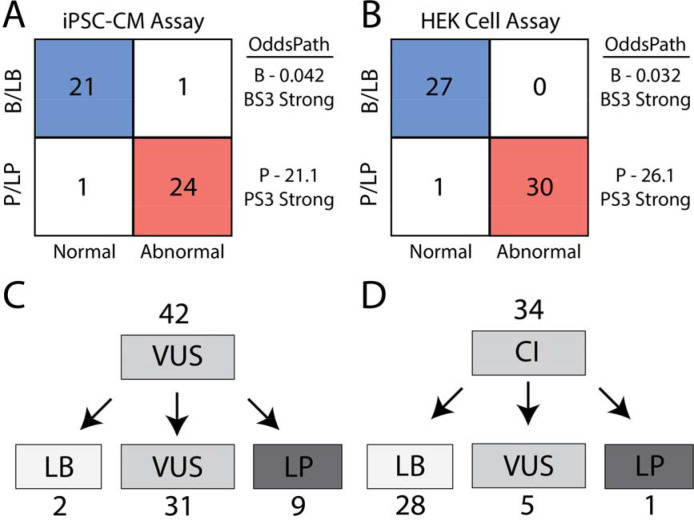
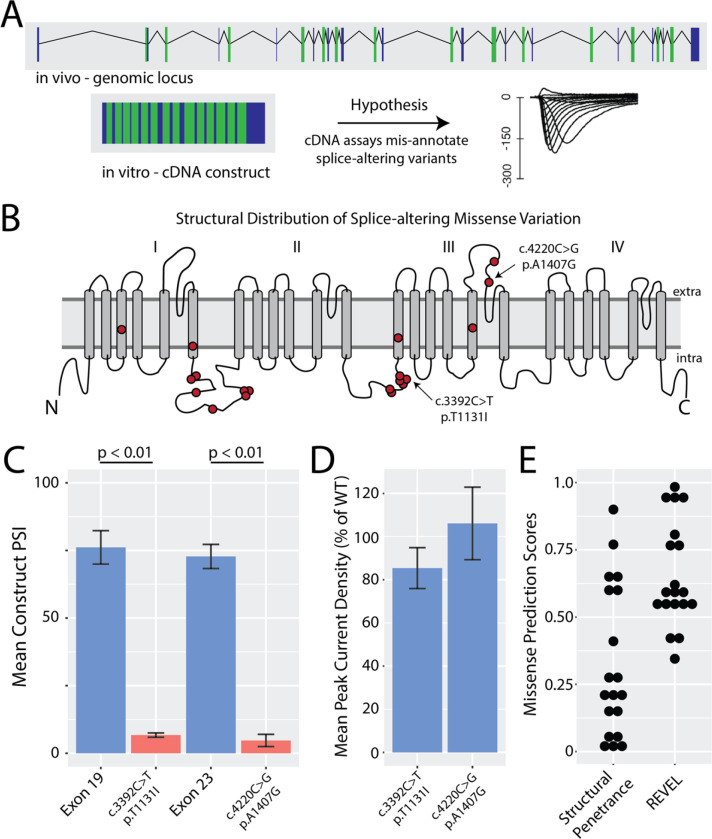
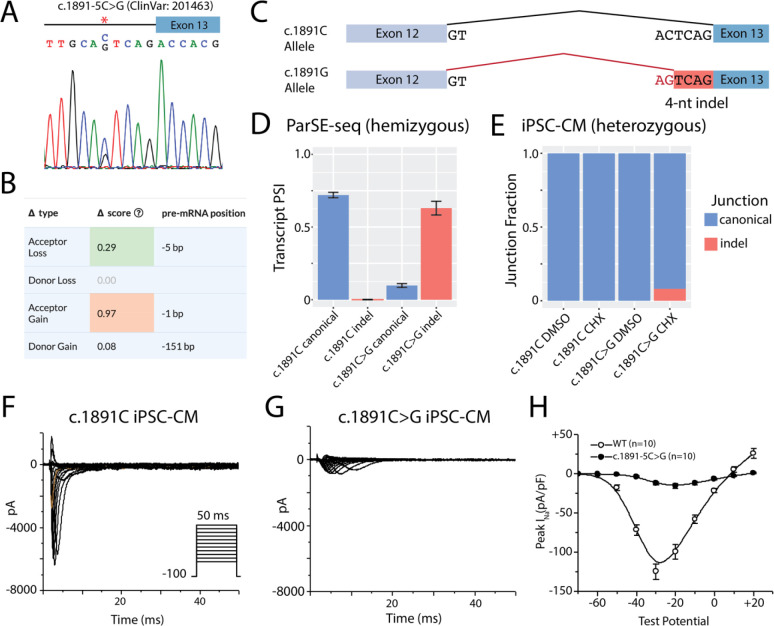
Results: ParSE-seq revealed the splicing effect of 224 SCN5A variants in iPSC-CMs and 244 variants in HEK293 cells. The scores between the cell types were highly correlated (R2=0.84). In iPSCs, the assay had concordant scores for 21/22 benign/likely benign and 24/25 pathogenic/likely pathogenic control variants from ClinVar. 43/112 exonic variants and 35/70 intronic variants with determinate scores disrupted splicing. 11 of 42 variants of uncertain significance were reclassified, and 29 of 34 variants with conflicting interpretations were reclassified using the functional data. SpliceAI computational predictions correlated well with experimental data (AUC = 0.96). We identified 20 unique SCN5A missense variants that disrupted splicing, and 2 clinically observed splice-altering missense variants of uncertain significance had normal function when tested with the cDNA-based APC assay. A splice-altering intronic variant detected by ParSE-seq, c.1891-5C>G, also disrupted splicing and sodium current when introduced into iPSC-CMs at the endogenous locus by CRISPR editing.
Conclusions: ParSE-seq is a calibrated, multiplexed, high-throughput assay to facilitate the classification of candidate splice-altering variants.
Keywords: Brugada syndrome; RNA; SCN5A; Splicing; arrhythmias; clinical genetics; iPSC-CM; non-coding; variant classification.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous