

Glucocorticoids, their uses, sexual dimorphisms, and diseases: new concepts, mechanisms, and discoveries
- PMID: 37732829
- PMCID: PMC11281820
- DOI: 10.1152/physrev.00021.2023
Glucocorticoids, their uses, sexual dimorphisms, and diseases: new concepts, mechanisms, and discoveries
Abstract
The normal stress response in humans is governed by the hypothalamic-pituitary-adrenal (HPA) axis through heightened mechanisms during stress, raising blood levels of the glucocorticoid hormone cortisol. Glucocorticoids are quintessential compounds that balance the proper functioning of numerous systems in the mammalian body. They are also generated synthetically and are the preeminent therapy for inflammatory diseases. They act by binding to the nuclear receptor transcription factor glucocorticoid receptor (GR), which has two main isoforms (GRα and GRβ). Our classical understanding of glucocorticoid signaling is from the GRα isoform, which binds the hormone, whereas GRβ has no known ligands. With glucocorticoids being involved in many physiological and cellular processes, even small disruptions in their release via the HPA axis, or changes in GR isoform expression, can have dire ramifications on health. Long-term chronic glucocorticoid therapy can lead to a glucocorticoid-resistant state, and we deliberate how this impacts disease treatment. Chronic glucocorticoid treatment can lead to noticeable side effects such as weight gain, adiposity, diabetes, and others that we discuss in detail. There are sexually dimorphic responses to glucocorticoids, and women tend to have a more hyperresponsive HPA axis than men. This review summarizes our understanding of glucocorticoids and critically analyzes the GR isoforms and their beneficial and deleterious mechanisms and the sexual differences that cause a dichotomy in responses. We also discuss the future of glucocorticoid therapy and propose a new concept of dual GR isoform agonist and postulate why activating both isoforms may prevent glucocorticoid resistance.
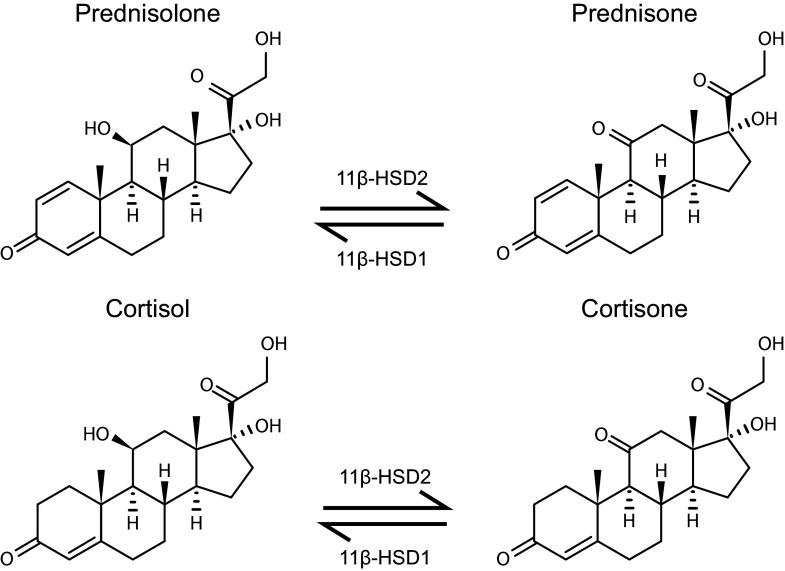
Keywords: 11β-HSD1; HPA axis; dual GR agonist; glucocorticoid resistance; inflammation.
Conflict of interest statement
T.D.H. has submitted patents on GRβ- and obesity-related disorders. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
Figures















References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical