

Coenzyme Q10 trapping in mitochondrial complex I underlies Leber's hereditary optic neuropathy
- PMID: 37733737
- PMCID: PMC10523484
- DOI: 10.1073/pnas.2304884120
Coenzyme Q10 trapping in mitochondrial complex I underlies Leber's hereditary optic neuropathy
Abstract
How does a single amino acid mutation occurring in the blinding disease, Leber's hereditary optic neuropathy (LHON), impair electron shuttling in mitochondria? We investigated changes induced by the m.3460 G>A mutation in mitochondrial protein ND1 using the tools of Molecular Dynamics and Free Energy Perturbation simulations, with the goal of determining the mechanism by which this mutation affects mitochondrial function. A recent analysis suggested that the mutation's replacement of alanine A52 with a threonine perturbs the stability of a region where binding of the electron shuttling protein, Coenzyme Q10, occurs. We found two functionally opposing changes involving the role of Coenzyme Q10. The first showed that quantum electron transfer from the terminal Fe/S complex, N2, to the Coenzyme Q10 headgroup, docked in its binding pocket, is enhanced. However, this positive adjustment is overshadowed by our finding that the mobility of Coenzyme Q10 in its oxidized and reduced states, entering and exiting its binding pocket, is disrupted by the mutation in a manner that leads to conditions promoting the generation of reactive oxygen species. An increase in reactive oxygen species caused by the LHON mutation has been proposed to be responsible for this optic neuropathy.
Keywords: Coenzyme Q10; blinding genetic disease; mitochondria; molecular dynamics simulation; quantum electron tunneling.
Conflict of interest statement
The authors declare no competing interest.
Figures
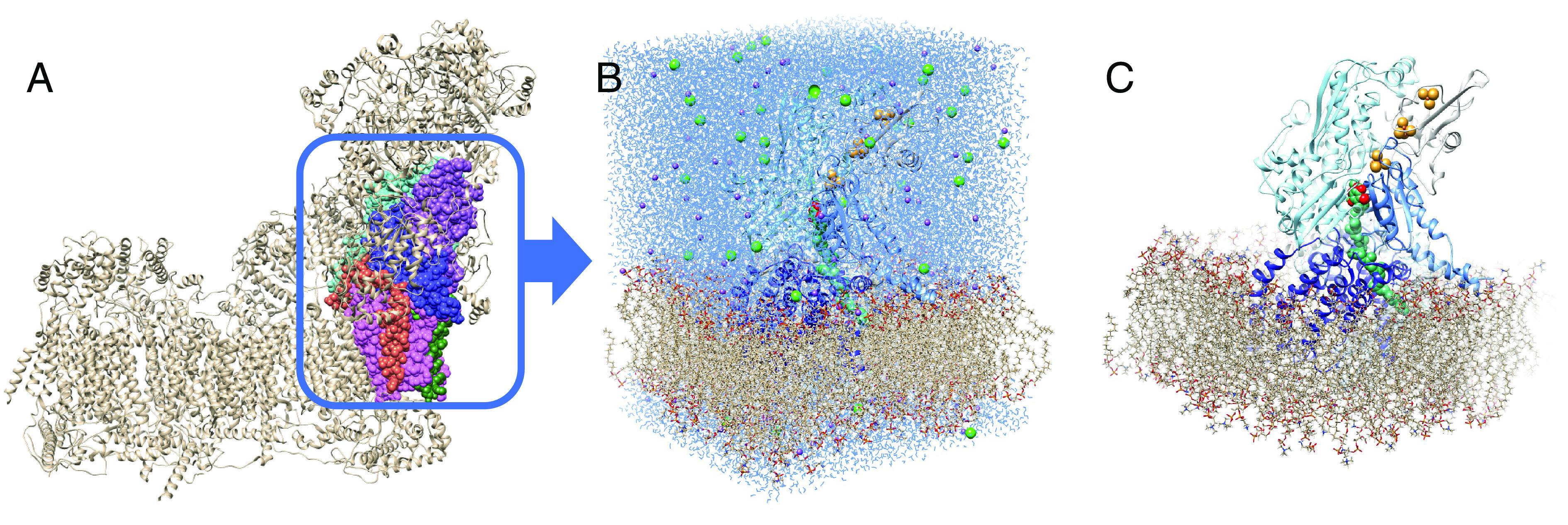
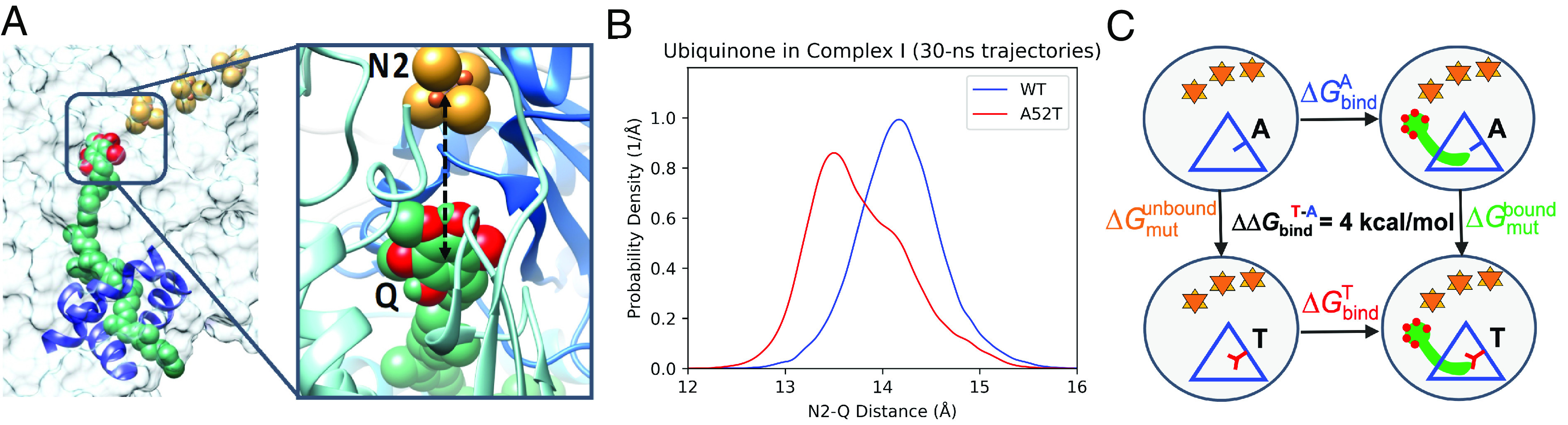
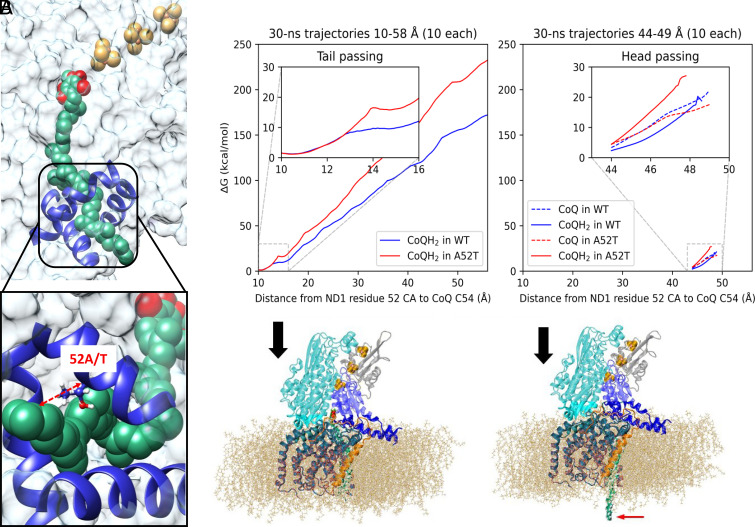
Comment in
-
How defective mitochondrial electrical activity leads to inherited blindness.Proc Natl Acad Sci U S A. 2023 Nov 7;120(45):e2315649120. doi: 10.1073/pnas.2315649120. Epub 2023 Oct 25. Proc Natl Acad Sci U S A. 2023. PMID: 37878684 Free PMC article. No abstract available.
Similar articles
-
The metabolomic signature of Leber's hereditary optic neuropathy reveals endoplasmic reticulum stress.Brain. 2016 Nov 1;139(11):2864-2876. doi: 10.1093/brain/aww222. Brain. 2016. PMID: 27633772
-
Leber's hereditary optic neuropathy caused by a mutation in mitochondrial tRNAThr in eight Chinese pedigrees.Mitochondrion. 2018 Sep;42:84-91. doi: 10.1016/j.mito.2017.12.003. Epub 2017 Dec 7. Mitochondrion. 2018. PMID: 29225014
-
Leber's Hereditary Optic Neuropathy as a Promising Disease for Gene Therapy Development.Adv Ther. 2019 Dec;36(12):3299-3307. doi: 10.1007/s12325-019-01113-2. Epub 2019 Oct 11. Adv Ther. 2019. PMID: 31605306 Free PMC article.
-
Leber's hereditary optic neuropathy-associated ND6 14484T > C mutation caused pleiotropic effects on the complex I, RNA homeostasis, apoptosis and mitophagy.Hum Mol Genet. 2022 Sep 29;31(19):3299-3312. doi: 10.1093/hmg/ddac109. Hum Mol Genet. 2022. PMID: 35567411
-
Mesenchymal stem cells (MSCs) in Leber's hereditary optic neuropathy (LHON): a potential therapeutic approach for future.Int Ophthalmol. 2022 Sep;42(9):2949-2964. doi: 10.1007/s10792-022-02267-9. Epub 2022 Mar 31. Int Ophthalmol. 2022. PMID: 35357640 Review.
Cited by
-
Mitochondrial diseases: from molecular mechanisms to therapeutic advances.Signal Transduct Target Ther. 2025 Jan 10;10(1):9. doi: 10.1038/s41392-024-02044-3. Signal Transduct Target Ther. 2025. PMID: 39788934 Free PMC article. Review.
-
How defective mitochondrial electrical activity leads to inherited blindness.Proc Natl Acad Sci U S A. 2023 Nov 7;120(45):e2315649120. doi: 10.1073/pnas.2315649120. Epub 2023 Oct 25. Proc Natl Acad Sci U S A. 2023. PMID: 37878684 Free PMC article. No abstract available.
-
Genetic variants affecting NQO1 protein levels impact the efficacy of idebenone treatment in Leber hereditary optic neuropathy.Cell Rep Med. 2024 Feb 20;5(2):101383. doi: 10.1016/j.xcrm.2023.101383. Epub 2024 Jan 24. Cell Rep Med. 2024. PMID: 38272025 Free PMC article.
-
Metformin may alter the course of Leber's hereditary optic neuropathy: a case report.Front Med (Lausanne). 2025 Aug 19;12:1609941. doi: 10.3389/fmed.2025.1609941. eCollection 2025. Front Med (Lausanne). 2025. PMID: 40904364 Free PMC article.
-
The Optic Nerve at Stake: Update on Environmental Factors Modulating Expression of Leber's Hereditary Optic Neuropathy.Biomedicines. 2024 Mar 6;12(3):584. doi: 10.3390/biomedicines12030584. Biomedicines. 2024. PMID: 38540197 Free PMC article. Review.
References
-
- Carelli V., “Leber’s hereditary optic neuropathy” in Mitochondrial Disorders in Neurology, Schapira A. H. V., DiMauro S., Eds. (Butterworth-Heinemann, 2002), pp. 115–142.
-
- Carelli V., Ross-Cisneros F. N., Sadun A. A., Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye. Res. 23, 53–89 (2004). - PubMed
-
- Wallace D. C., et al. , Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242, 1427–1430 (1988). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
