

ATM Mutations Associate with Distinct Co-Mutational Patterns and Therapeutic Vulnerabilities in NSCLC
- PMID: 37733794
- PMCID: PMC10690143
- DOI: 10.1158/1078-0432.CCR-23-1122
ATM Mutations Associate with Distinct Co-Mutational Patterns and Therapeutic Vulnerabilities in NSCLC
Abstract
Purpose: Ataxia-telangiectasia mutated (ATM) is the most frequently mutated DNA damage repair gene in non-small cell lung cancer (NSCLC). However, the molecular correlates of ATM mutations and their clinical implications have not been fully elucidated.
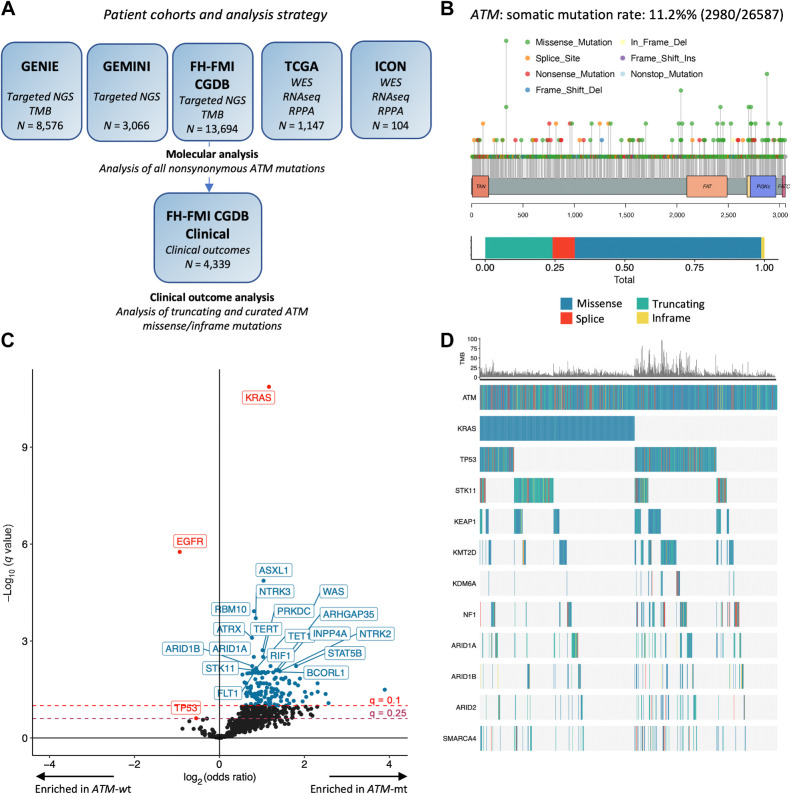
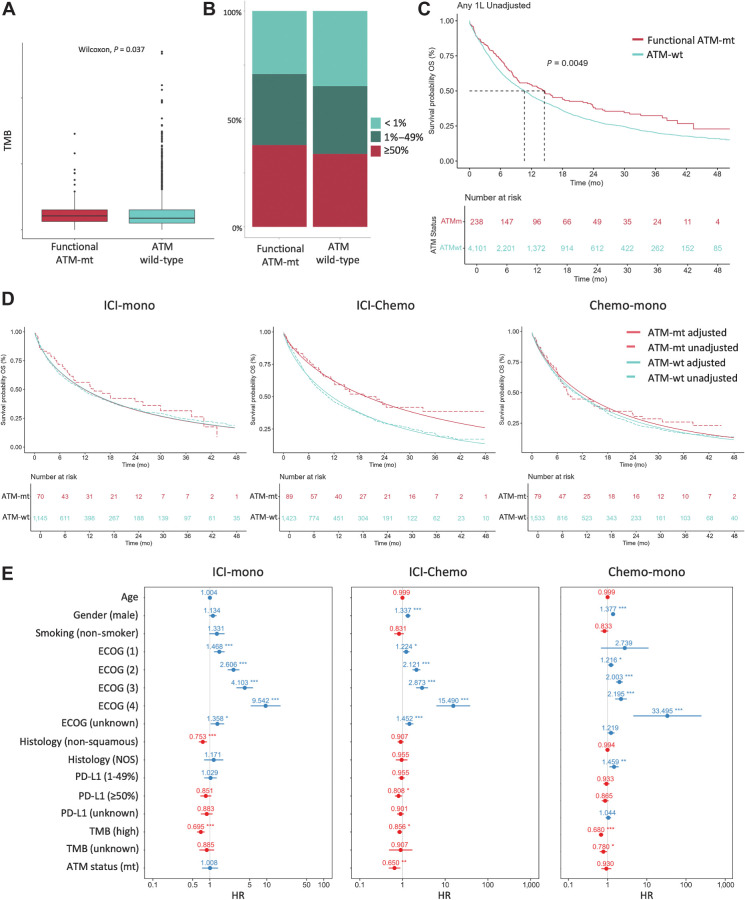
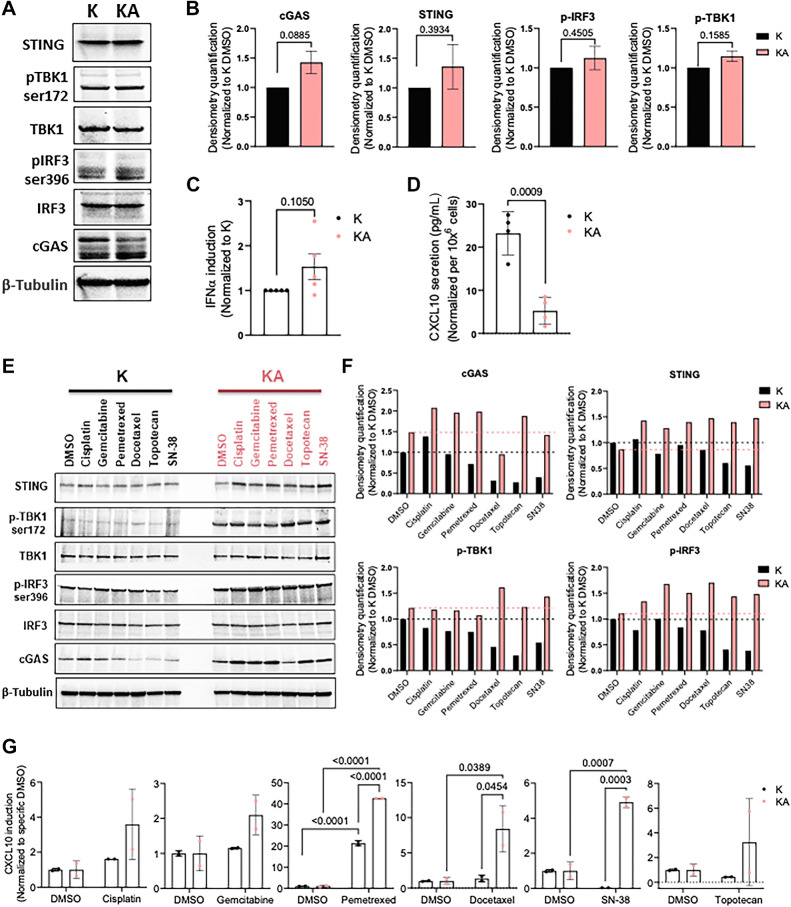
Experimental design: Clinicopathologic and genomic data from 26,587 patients with NSCLC from MD Anderson, public databases, and a de-identified nationwide (US-based) NSCLC clinicogenomic database (CGDB) were used to assess the co-mutation landscape, protein expression, and mutational processes in ATM-mutant tumors. We used the CGDB to evaluate ATM-associated outcomes in patients treated with immune checkpoint inhibitors (ICI) with or without chemotherapy, and assessed the effect of ATM loss on STING signaling and chemotherapy sensitivity in preclinical models.
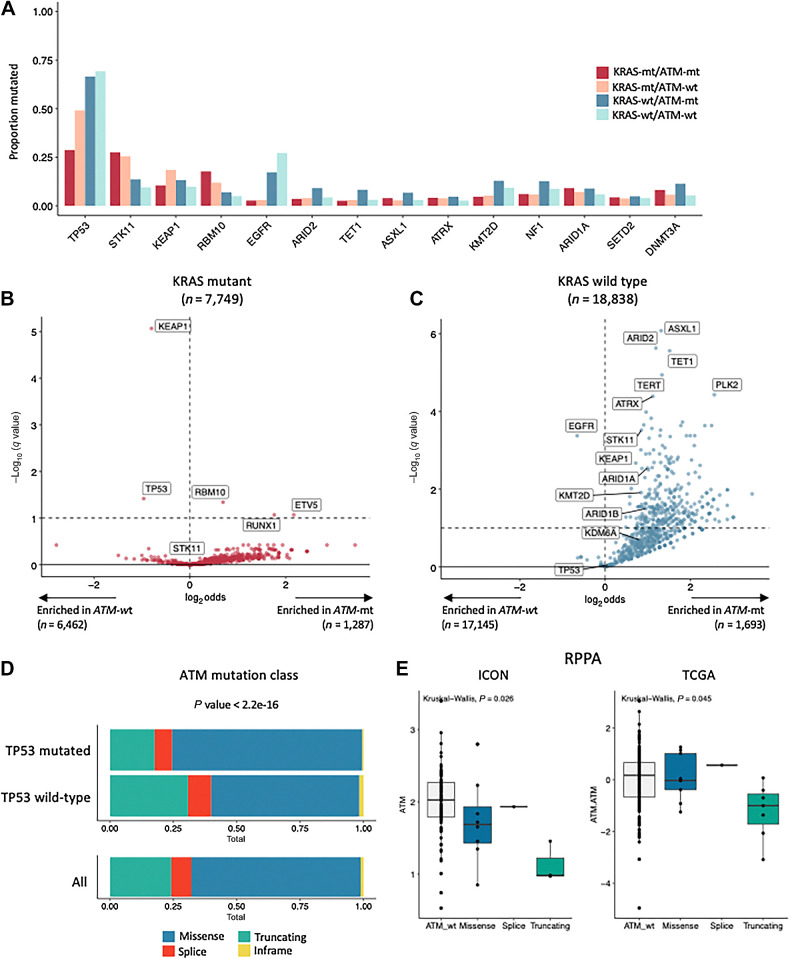
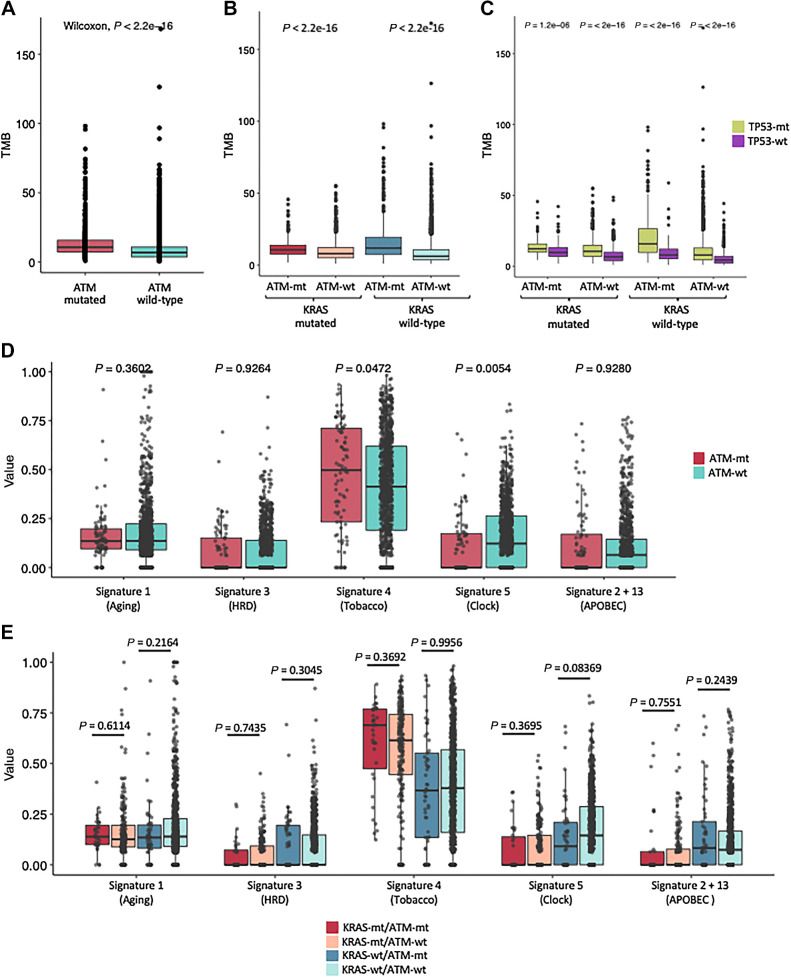
Results: Nonsynonymous mutations in ATM were observed in 11.2% of samples (2,980/26,587) and were significantly associated with mutations in KRAS, but mutually exclusive with EGFR (q < 0.1). KRAS mutational status constrained the ATM co-mutation landscape, with strong mutual exclusivity with TP53 and KEAP1 within KRAS-mutated samples. Those ATM mutations that co-occurred with TP53 were more likely to be missense mutations and associate with high mutational burden, suggestive of non-functional passenger mutations. In the CGDB cohort, dysfunctional ATM mutations associated with improved OS only in patients treated with ICI-chemotherapy, and not ICI alone. In vitro analyses demonstrated enhanced upregulation of STING signaling in ATM knockout cells with the addition of chemotherapy.
Conclusions: ATM mutations define a distinct subset of NSCLC associated with KRAS mutations, increased TMB, decreased TP53 and EGFR co-occurrence, and potential increased sensitivity to ICIs in the context of DNA-damaging chemotherapy.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
Similar articles
-
Association of Tumor Protein p53 and Ataxia-Telangiectasia Mutated Comutation With Response to Immune Checkpoint Inhibitors and Mortality in Patients With Non-Small Cell Lung Cancer.JAMA Netw Open. 2019 Sep 4;2(9):e1911895. doi: 10.1001/jamanetworkopen.2019.11895. JAMA Netw Open. 2019. PMID: 31539077 Free PMC article.
-
The prevalence and real-world therapeutic analysis of Chinese patients with KRAS-Mutant Non-Small Cell lung cancer.Cancer Med. 2022 Oct;11(19):3581-3592. doi: 10.1002/cam4.4739. Epub 2022 Apr 8. Cancer Med. 2022. PMID: 35394121 Free PMC article.
-
Clinicopathologic, Genomic, and Immunophenotypic Landscape of ATM Mutations in Non-Small Cell Lung Cancer.Clin Cancer Res. 2023 Jul 5;29(13):2540-2550. doi: 10.1158/1078-0432.CCR-22-3413. Clin Cancer Res. 2023. PMID: 37097610 Free PMC article.
-
To be, or not to be: the dilemma of immunotherapy for non-small cell lung cancer harboring various driver mutations.J Cancer Res Clin Oncol. 2023 Sep;149(12):10027-10040. doi: 10.1007/s00432-023-04919-4. Epub 2023 Jun 1. J Cancer Res Clin Oncol. 2023. PMID: 37261523 Free PMC article. Review.
-
KRAS G12C in advanced NSCLC: Prevalence, co-mutations, and testing.Lung Cancer. 2023 Oct;184:107293. doi: 10.1016/j.lungcan.2023.107293. Epub 2023 Jul 13. Lung Cancer. 2023. PMID: 37683526 Review.
Cited by
-
Novel immune checkpoint inhibitor strategies in advanced non-small cell lung cancer: towards biomarker-driven therapies?Transl Lung Cancer Res. 2025 Feb 28;14(2):328-333. doi: 10.21037/tlcr-24-966. Epub 2025 Feb 24. Transl Lung Cancer Res. 2025. PMID: 40114950 Free PMC article. No abstract available.
-
Precision immune regulation in KRAS-mutated cancers: the final piece of the puzzle?J Exp Clin Cancer Res. 2025 Jul 3;44(1):189. doi: 10.1186/s13046-025-03444-1. J Exp Clin Cancer Res. 2025. PMID: 40611261 Free PMC article. Review.
-
The Prognostic and Predictive Roles of Ataxia-Telangiectasia Mutated (ATM) Expression in Patients with Metastatic Non-Small-Cell Lung Cancer Receiving Pembrolizumab Monotherapy Alone or in Combination with Chemotherapy.Diagnostics (Basel). 2025 Apr 21;15(8):1048. doi: 10.3390/diagnostics15081048. Diagnostics (Basel). 2025. PMID: 40310425 Free PMC article.
-
Co-Occurring Genomic Alterations in NSCLC: Making Order into a Crowded List.Cancers (Basel). 2025 Jul 18;17(14):2388. doi: 10.3390/cancers17142388. Cancers (Basel). 2025. PMID: 40723270 Free PMC article. Review.
-
PARP inhibitor synthetic lethality in ATM biallelic mutant cancer cell lines is associated with BRCA1/2 and RAD51 downregulation.Front Oncol. 2024 May 14;14:1380633. doi: 10.3389/fonc.2024.1380633. eCollection 2024. Front Oncol. 2024. PMID: 38807759 Free PMC article.
References
-
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008;9:759–69. - PubMed
-
- Taylor AMR, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, et al. . Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature 1975;258:427–9. - PubMed
-
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol 1997;15:177–202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
