

The arginase 1/ornithine decarboxylase pathway suppresses HDAC3 to ameliorate the myeloid cell inflammatory response: implications for retinal ischemic injury
- PMID: 37735154
- PMCID: PMC10514323
- DOI: 10.1038/s41419-023-06147-7
The arginase 1/ornithine decarboxylase pathway suppresses HDAC3 to ameliorate the myeloid cell inflammatory response: implications for retinal ischemic injury
Abstract
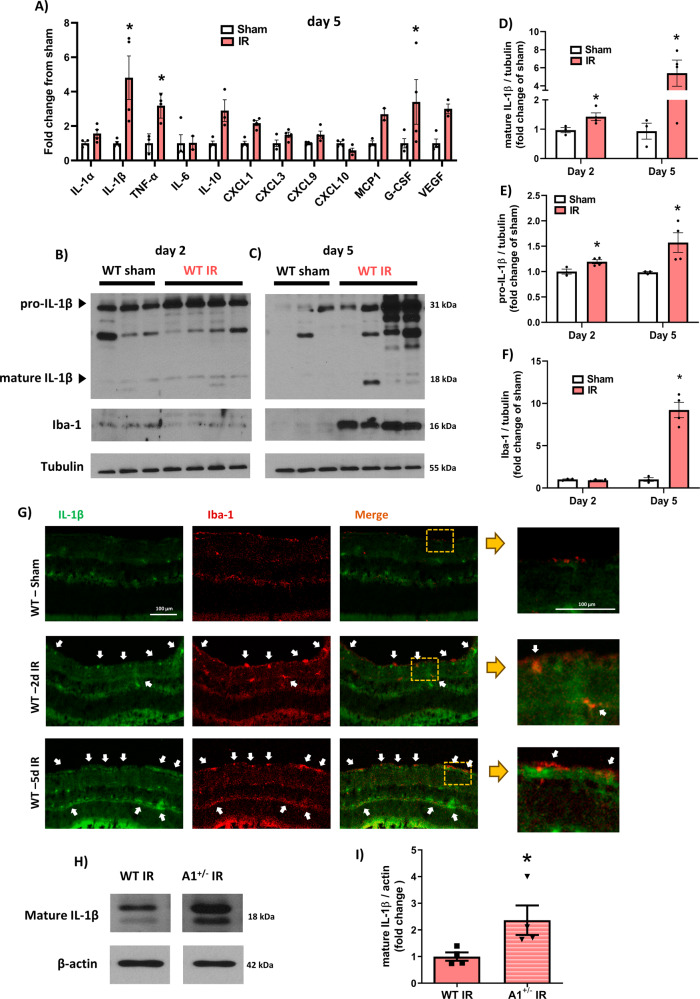
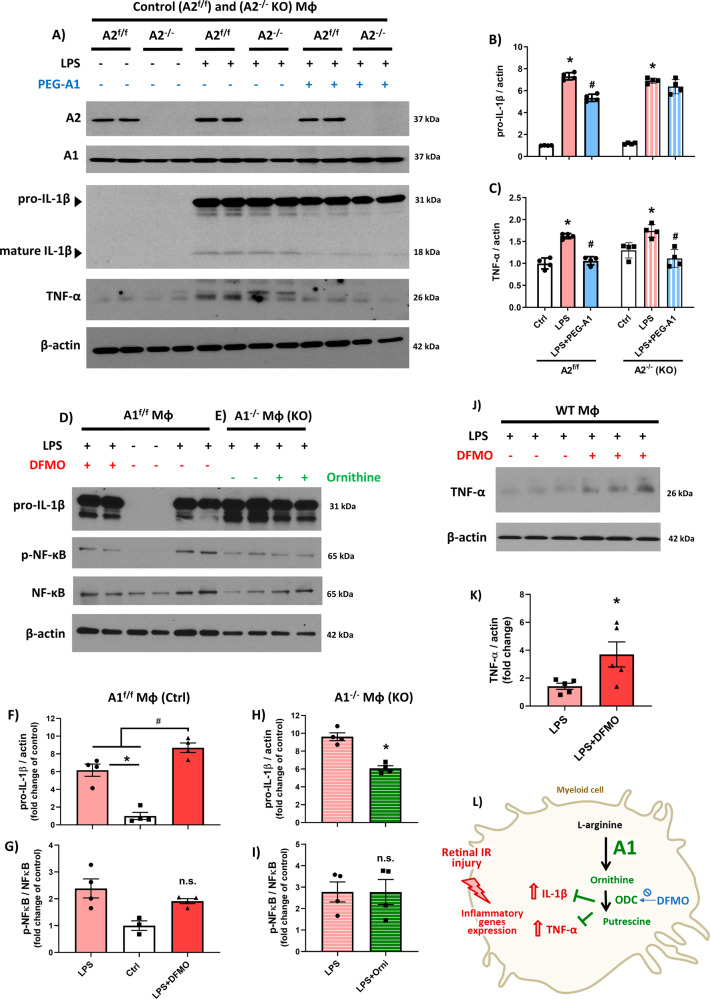
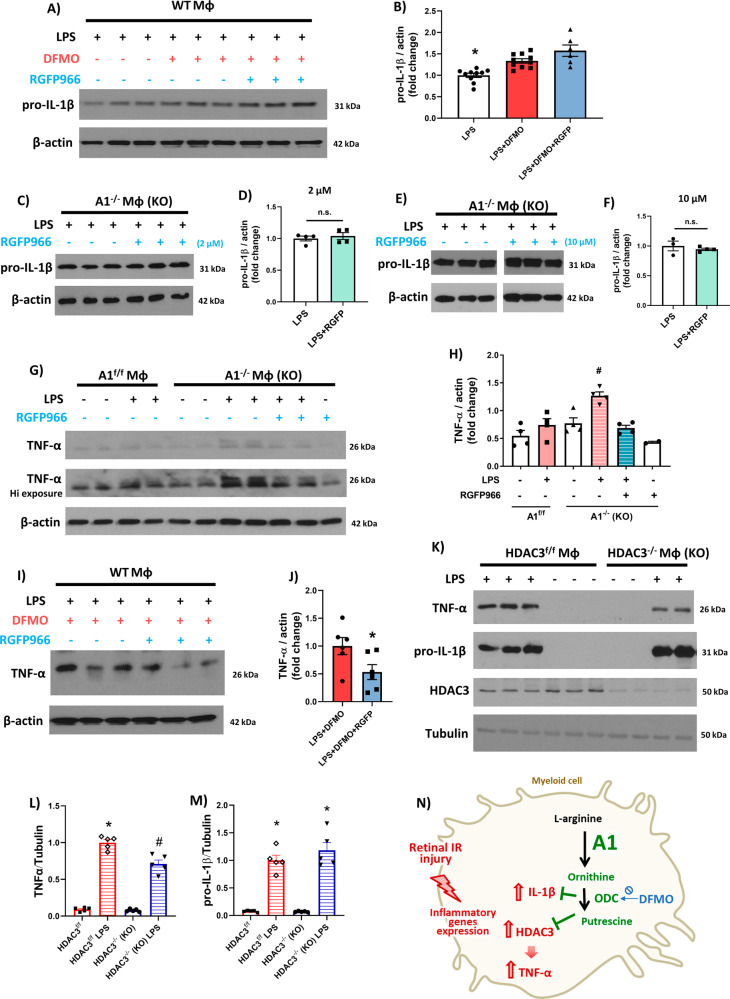
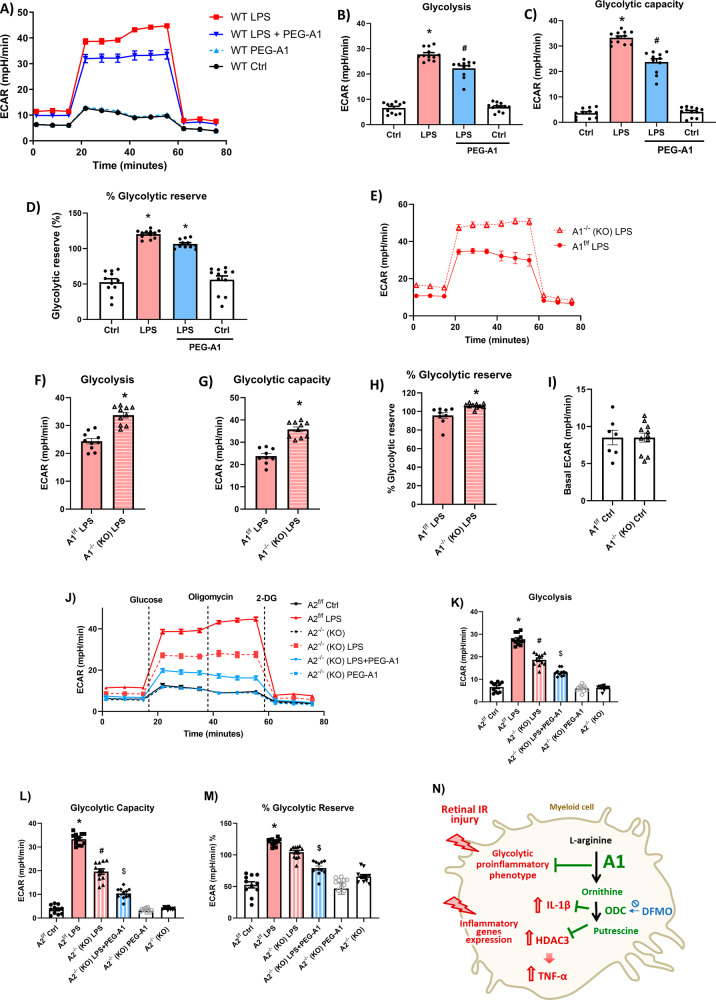
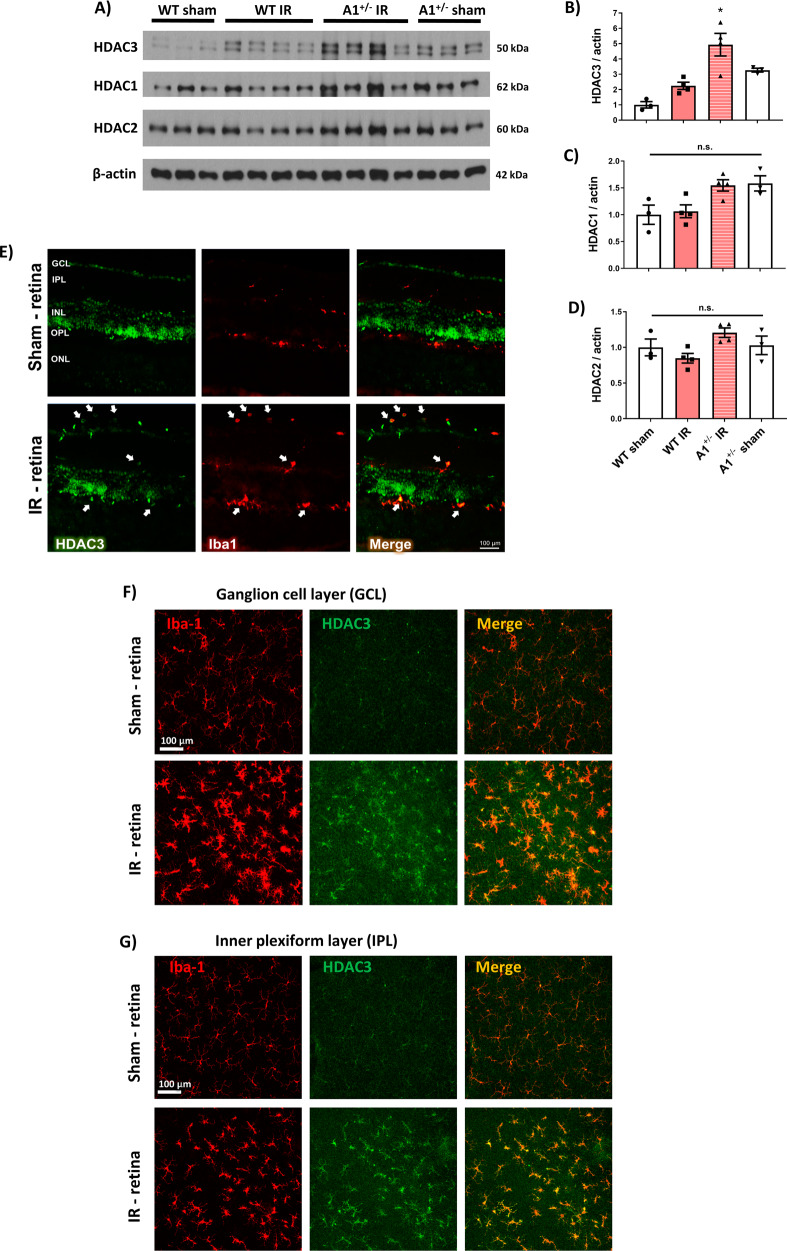
The enzyme arginase 1 (A1) hydrolyzes the amino acid arginine to form L-ornithine and urea. Ornithine is further converted to polyamines by the ornithine decarboxylase (ODC) enzyme. We previously reported that deletion of myeloid A1 in mice exacerbates retinal damage after ischemia/reperfusion (IR) injury. Furthermore, treatment with A1 protects against retinal IR injury in wild-type mice. PEG-A1 also mitigates the exaggerated inflammatory response of A1 knockout (KO) macrophages in vitro. Here, we sought to identify the anti-inflammatory pathway that confers macrophage A1-mediated protection against retinal IR injury. Acute elevation of intraocular pressure was used to induce retinal IR injury in mice. A multiplex cytokine assay revealed a marked increase in the inflammatory cytokines interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α) in the retina at day 5 after IR injury. In vitro, blocking the A1/ODC pathway augmented IL-1β and TNF-α production in stimulated macrophages. Furthermore, A1 treatment attenuated the stimulated macrophage metabolic switch to a pro-inflammatory glycolytic phenotype, whereas A1 deletion had the opposite effect. Screening for histone deacetylases (HDACs) which play a role in macrophage inflammatory response showed that A1 deletion or ODC inhibition increased the expression of HDAC3. We further showed the involvement of HDAC3 in the upregulation of TNF-α but not IL-1β in stimulated macrophages deficient in the A1/ODC pathway. Investigating HDAC3 KO macrophages showed a reduced inflammatory response and a less glycolytic phenotype upon stimulation. In vivo, HDAC3 co-localized with microglia/macrophages at day 2 after IR in WT retinas and was further increased in A1-deficient retinas. Collectively, our data provide initial evidence that A1 exerts its anti-inflammatory effect in macrophages via ODC-mediated suppression of HDAC3 and IL-1β. Collectively we propose that interventions that augment the A1/ODC pathway and inhibit HDAC3 may confer therapeutic benefits for the treatment of retinal ischemic diseases.
© 2023. The Author(s).
Conflict of interest statement
AYF and RBC have a pending patent on the use of arginase 1 as a treatment for ischemic retinopathies. The other authors declare no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
