

Identification of mobile genetic elements with geNomad
- PMID: 37735266
- PMCID: PMC11324519
- DOI: 10.1038/s41587-023-01953-y
Identification of mobile genetic elements with geNomad
Abstract
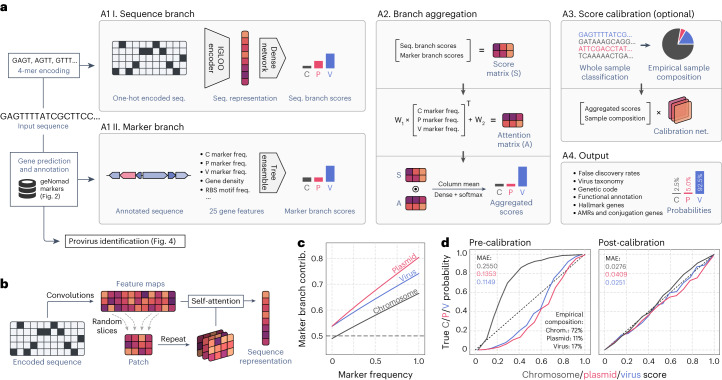
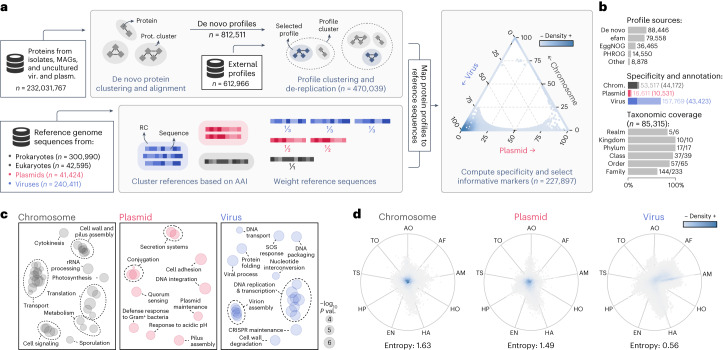
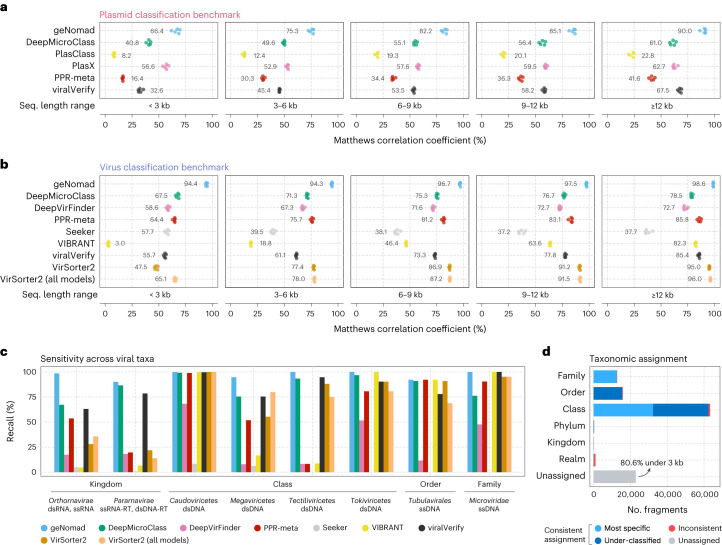
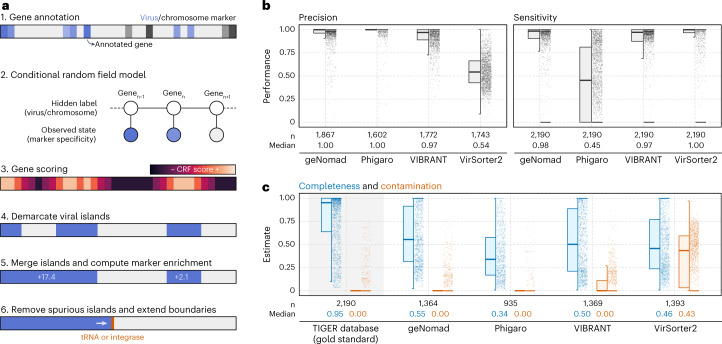
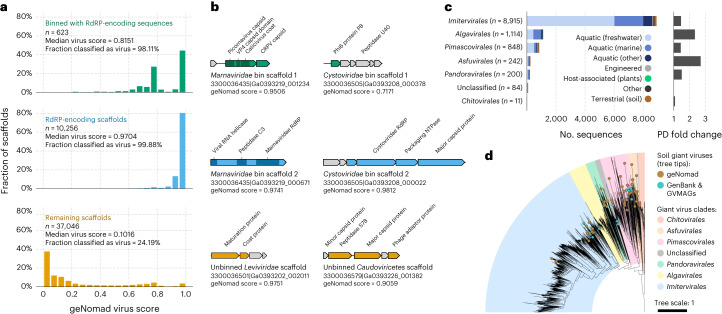
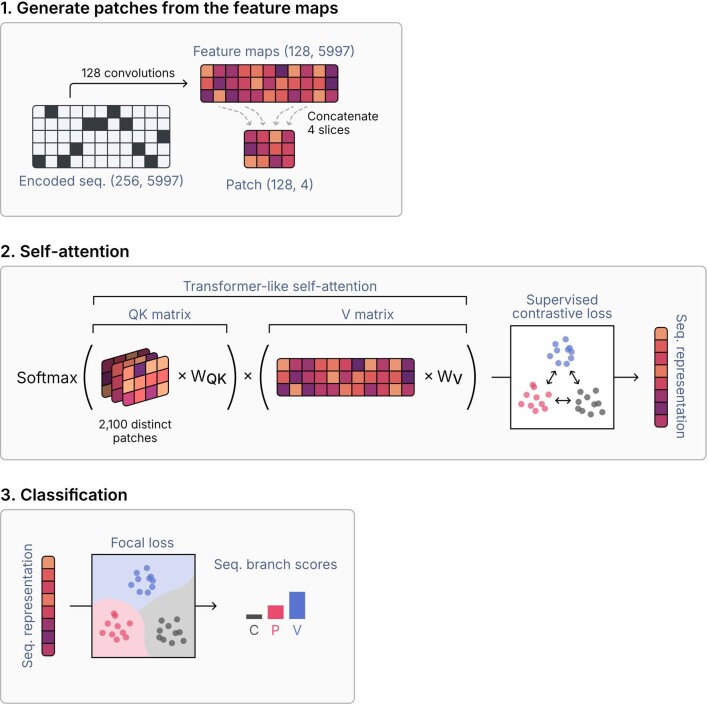
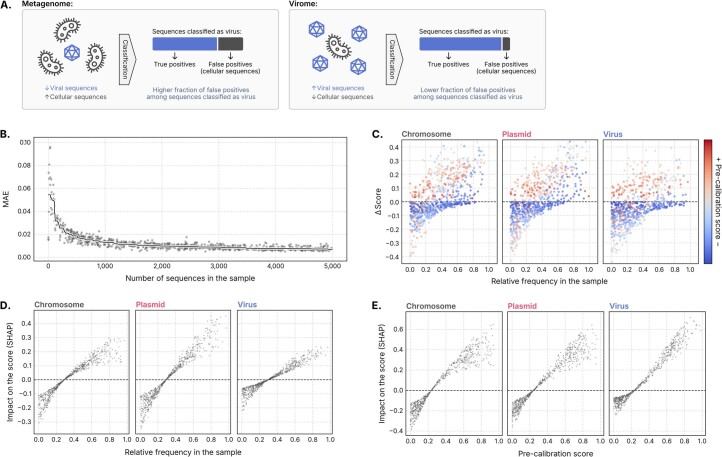
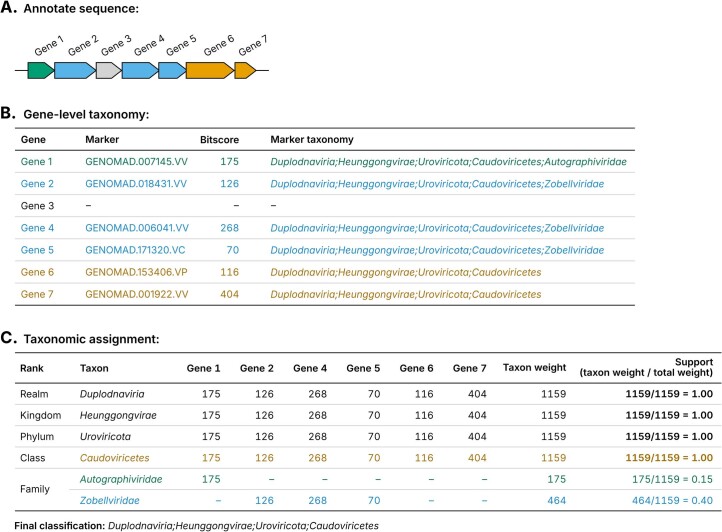
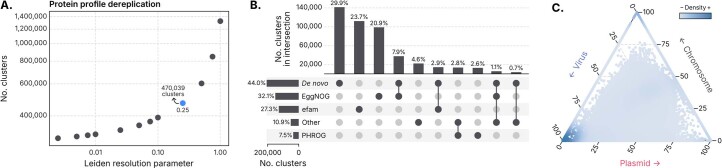
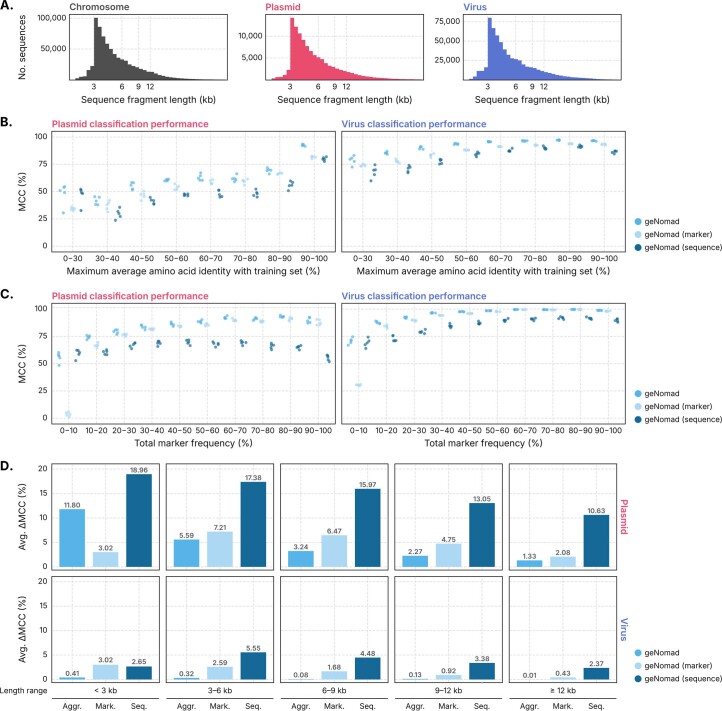
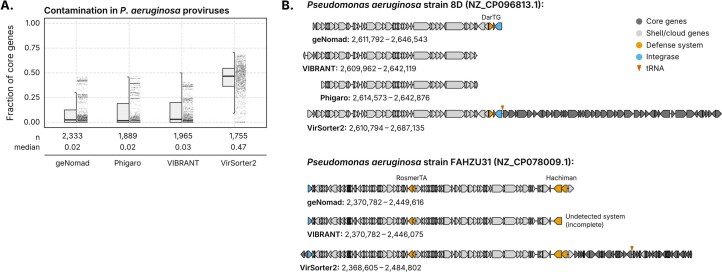
Identifying and characterizing mobile genetic elements in sequencing data is essential for understanding their diversity, ecology, biotechnological applications and impact on public health. Here we introduce geNomad, a classification and annotation framework that combines information from gene content and a deep neural network to identify sequences of plasmids and viruses. geNomad uses a dataset of more than 200,000 marker protein profiles to provide functional gene annotation and taxonomic assignment of viral genomes. Using a conditional random field model, geNomad also detects proviruses integrated into host genomes with high precision. In benchmarks, geNomad achieved high classification performance for diverse plasmids and viruses (Matthews correlation coefficient of 77.8% and 95.3%, respectively), substantially outperforming other tools. Leveraging geNomad's speed and scalability, we processed over 2.7 trillion base pairs of sequencing data, leading to the discovery of millions of viruses and plasmids that are available through the IMG/VR and IMG/PR databases. geNomad is available at https://portal.nersc.gov/genomad .
© 2023. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata.Nucleic Acids Res. 2023 Jan 6;51(D1):D733-D743. doi: 10.1093/nar/gkac1037. Nucleic Acids Res. 2023. PMID: 36399502 Free PMC article.
-
MVP: a modular viromics pipeline to identify, filter, cluster, annotate, and bin viruses from metagenomes.mSystems. 2024 Oct 22;9(10):e0088824. doi: 10.1128/msystems.00888-24. Epub 2024 Oct 1. mSystems. 2024. PMID: 39352141 Free PMC article.
-
IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses.Nucleic Acids Res. 2021 Jan 8;49(D1):D764-D775. doi: 10.1093/nar/gkaa946. Nucleic Acids Res. 2021. PMID: 33137183 Free PMC article.
-
A host of armor: Prokaryotic immune strategies against mobile genetic elements.Cell Rep. 2023 Jul 25;42(7):112672. doi: 10.1016/j.celrep.2023.112672. Epub 2023 Jun 21. Cell Rep. 2023. PMID: 37347666 Review.
-
Horizontal gene transfer and mobile genetic elements in marine systems.Methods Mol Biol. 2009;532:435-53. doi: 10.1007/978-1-60327-853-9_25. Methods Mol Biol. 2009. PMID: 19271200 Review.
Cited by
-
Phylogenomic analysis expands the known repertoire of single-stranded DNA viruses in benthic zones of the South Indian Ocean.ISME Commun. 2024 May 1;4(1):ycae065. doi: 10.1093/ismeco/ycae065. eCollection 2024 Jan. ISME Commun. 2024. PMID: 38800127 Free PMC article.
-
Isolation of phages infecting the zoonotic pathogen Streptococcus suis reveals novel structural and genomic characteristics.bioRxiv [Preprint]. 2025 Jan 7:2025.01.07.631744. doi: 10.1101/2025.01.07.631744. bioRxiv. 2025. Update in: Microbiol Res. 2025 Jul;296:128147. doi: 10.1016/j.micres.2025.128147. PMID: 39829746 Free PMC article. Updated. Preprint.
-
Protein Set Transformer: A protein-based genome language model to power high diversity viromics.Res Sq [Preprint]. 2024 Sep 23:rs.3.rs-4844047. doi: 10.21203/rs.3.rs-4844047/v1. Res Sq. 2024. PMID: 39399683 Free PMC article. Preprint.
-
Genomic and morphological features of an Amazonian Bacillus thuringiensis with mosquito larvicidal activity.AMB Express. 2025 Mar 5;15(1):39. doi: 10.1186/s13568-025-01850-4. AMB Express. 2025. PMID: 40045023 Free PMC article.
-
Metagenomics Disentangles Differential Resistome Traits and Risks in Full-Scale Anaerobic Digestion Plants under Ambient, Mesophilic, and Thermophilic Conditions.ACS Environ Au. 2024 Dec 5;5(2):183-196. doi: 10.1021/acsenvironau.4c00071. eCollection 2025 Mar 19. ACS Environ Au. 2024. PMID: 40125276 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
