

Critical assessment of on-premise approaches to scalable genome analysis
- PMID: 37735350
- PMCID: PMC10512525
- DOI: 10.1186/s12859-023-05470-2
Critical assessment of on-premise approaches to scalable genome analysis
Abstract
Background: Plummeting DNA sequencing cost in recent years has enabled genome sequencing projects to scale up by several orders of magnitude, which is transforming genomics into a highly data-intensive field of research. This development provides the much needed statistical power required for genotype-phenotype predictions in complex diseases.
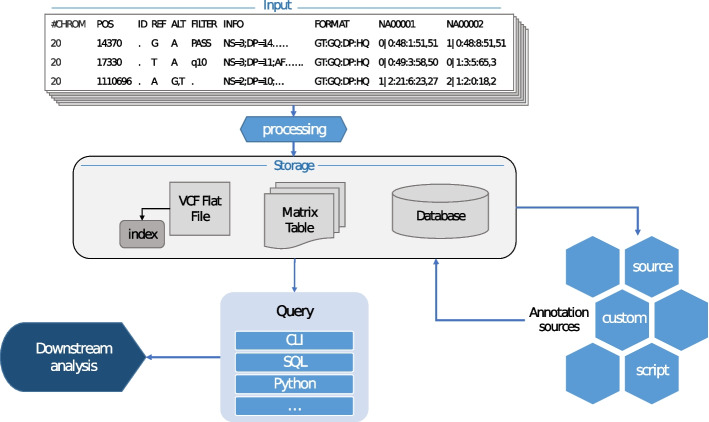
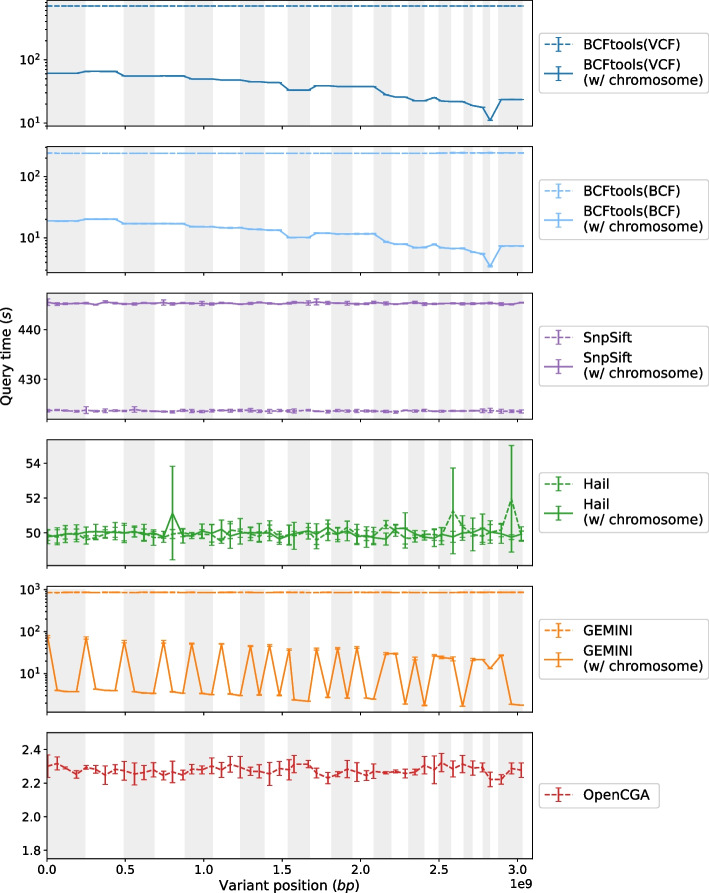
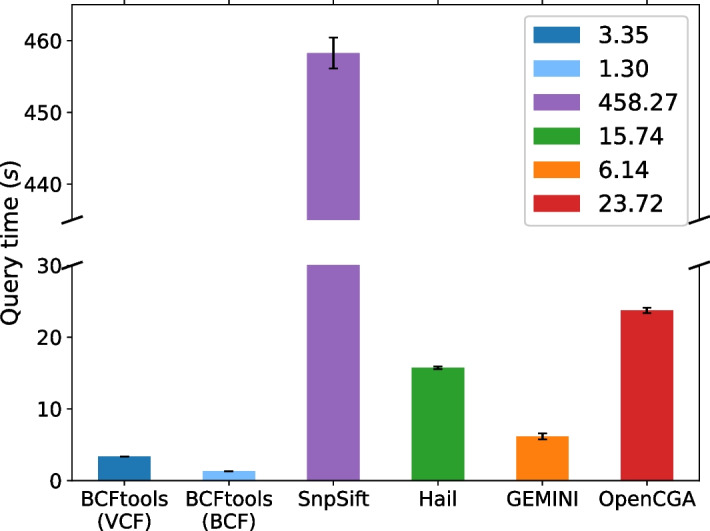
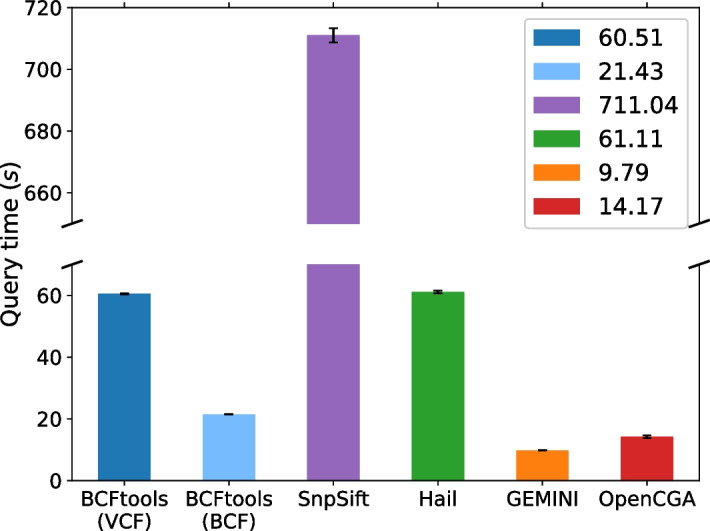
Methods: In order to efficiently leverage the wealth of information, we here assessed several genomic data science tools. The rationale to focus on on-premise installations is to cope with situations where data confidentiality and compliance regulations etc. rule out cloud based solutions. We established a comprehensive qualitative and quantitative comparison between BCFtools, SnpSift, Hail, GEMINI, and OpenCGA. The tools were compared in terms of data storage technology, query speed, scalability, annotation, data manipulation, visualization, data output representation, and availability.
Results: Tools that leverage sophisticated data structures are noted as the most suitable for large-scale projects in varying degrees of scalability in comparison to flat-file manipulation (e.g., BCFtools, and SnpSift). Remarkably, for small to mid-size projects, even lightweight relational database.
Conclusion: The assessment criteria provide insights into the typical questions posed in scalable genomics and serve as guidance for the development of scalable computational infrastructure in genomics.
Keywords: Big data; Genomic data science; Genomic databases; Horizontal scaling; NoSQL; SQL; VCF.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Shared data science infrastructure for genomics data.BMC Bioinformatics. 2019 Aug 22;20(1):436. doi: 10.1186/s12859-019-2967-2. BMC Bioinformatics. 2019. PMID: 31438850 Free PMC article.
-
SeqWare Query Engine: storing and searching sequence data in the cloud.BMC Bioinformatics. 2010 Dec 21;11 Suppl 12(Suppl 12):S2. doi: 10.1186/1471-2105-11-S12-S2. BMC Bioinformatics. 2010. PMID: 21210981 Free PMC article.
-
Evaluation of relational and NoSQL database architectures to manage genomic annotations.J Biomed Inform. 2016 Dec;64:288-295. doi: 10.1016/j.jbi.2016.10.015. Epub 2016 Oct 31. J Biomed Inform. 2016. PMID: 27810480
-
Trends in IT Innovation to Build a Next Generation Bioinformatics Solution to Manage and Analyse Biological Big Data Produced by NGS Technologies.Biomed Res Int. 2015;2015:904541. doi: 10.1155/2015/904541. Epub 2015 Jun 1. Biomed Res Int. 2015. PMID: 26125026 Free PMC article. Review.
-
Lessons learnt on the analysis of large sequence data in animal genomics.Anim Genet. 2018 Jun;49(3):147-158. doi: 10.1111/age.12655. Epub 2018 Apr 6. Anim Genet. 2018. PMID: 29624711 Review.
Cited by
-
Analysis-ready VCF at Biobank scale using Zarr.bioRxiv [Preprint]. 2025 Feb 6:2024.06.11.598241. doi: 10.1101/2024.06.11.598241. bioRxiv. 2025. Update in: Gigascience. 2025 Jan 6;14:giaf049. doi: 10.1093/gigascience/giaf049. PMID: 38915693 Free PMC article. Updated. Preprint.
-
Analysis-ready VCF at Biobank scale using Zarr.Gigascience. 2025 Jan 6;14:giaf049. doi: 10.1093/gigascience/giaf049. Gigascience. 2025. PMID: 40451243 Free PMC article.
References
-
- Ku CS, Loy EY, Salim A, Pawitan Y, Chia KS. The discovery of human genetic variations and their use as disease markers: past, present and future. J Hum Genet. 2010;55(7):403–415. - PubMed
-
- Chellappa SA, Pathak AK, Sinha P, Jainarayanan AK, Jain S, Brahmachari SK. Meta-analysis of genomic variants and gene expression data in schizophrenia suggests the potential need for adjunctive therapeutic interventions for neuropsychiatric disorders. J Genet. 2019;98(2):1–13. - PubMed
MeSH terms
Grants and funding
- CIRA-2019-076/Khalifa University of Science, Technology and Research
- CIRA-2019-076/Khalifa University of Science, Technology and Research
- CIRA-2019-076/Khalifa University of Science, Technology and Research
- CIRA-2019-076/Khalifa University of Science, Technology and Research
- CIRA-2019-076/Khalifa University of Science, Technology and Research
LinkOut - more resources
Full Text Sources
