

Deep generative modeling of transcriptional dynamics for RNA velocity analysis in single cells
- PMID: 37735568
- PMCID: PMC10776389
- DOI: 10.1038/s41592-023-01994-w
Deep generative modeling of transcriptional dynamics for RNA velocity analysis in single cells
Abstract
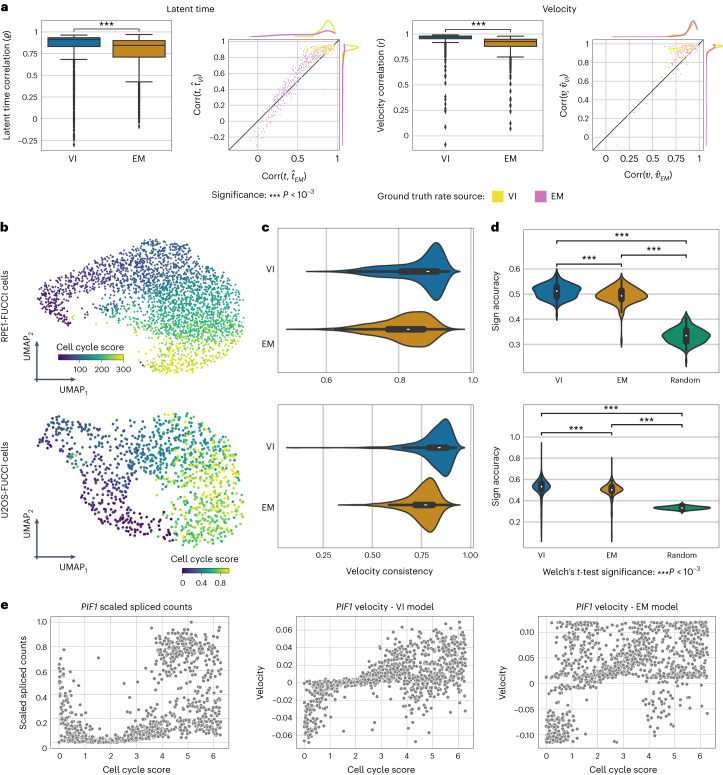
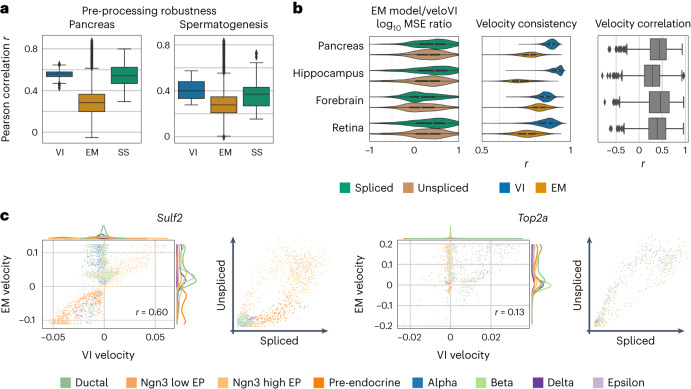
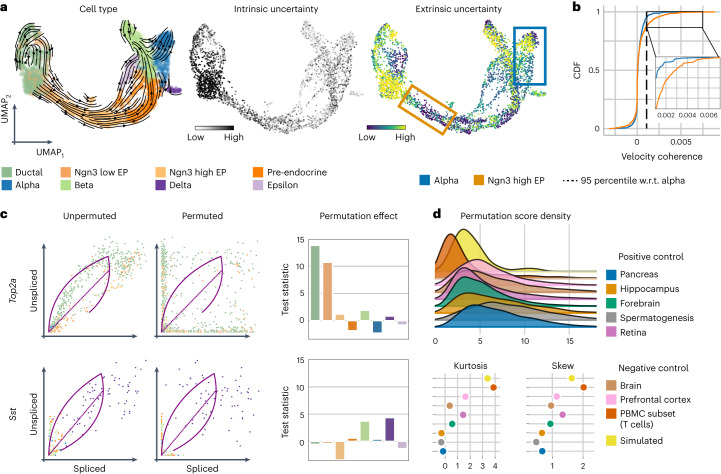
RNA velocity has been rapidly adopted to guide interpretation of transcriptional dynamics in snapshot single-cell data; however, current approaches for estimating RNA velocity lack effective strategies for quantifying uncertainty and determining the overall applicability to the system of interest. Here, we present veloVI (velocity variational inference), a deep generative modeling framework for estimating RNA velocity. veloVI learns a gene-specific dynamical model of RNA metabolism and provides a transcriptome-wide quantification of velocity uncertainty. We show that veloVI compares favorably to previous approaches with respect to goodness of fit, consistency across transcriptionally similar cells and stability across preprocessing pipelines for quantifying RNA abundance. Further, we demonstrate that veloVI's posterior velocity uncertainty can be used to assess whether velocity analysis is appropriate for a given dataset. Finally, we highlight veloVI as a flexible framework for modeling transcriptional dynamics by adapting the underlying dynamical model to use time-dependent transcription rates.
© 2023. The Author(s).
Conflict of interest statement
M.L. consults for Santa Ana Bio, is a part-time employee at Relation Therapeutics and owns interests in Relation Therapeutics. F.J.T. consults for Immunai, Singularity Bio, CytoReason and Omniscope and has ownership interest in Dermagnostix and Cellarity. N.Y. is an advisor and/or has equity in Cellarity, Celsius Therapeutics and Rheos Medicine. The remaining authors declare no competing interests.
Figures









Similar articles
-
Dynamical Systems Model of RNA Velocity Improves Inference of Single-cell Trajectory, Pseudo-time and Gene Regulation.J Mol Biol. 2022 Aug 15;434(15):167606. doi: 10.1016/j.jmb.2022.167606. Epub 2022 Apr 27. J Mol Biol. 2022. PMID: 35489382
-
DeepKINET: a deep generative model for estimating single-cell RNA splicing and degradation rates.Genome Biol. 2024 Sep 6;25(1):229. doi: 10.1186/s13059-024-03367-8. Genome Biol. 2024. PMID: 39237934 Free PMC article.
-
Reconstructing developmental trajectories using latent dynamical systems and time-resolved transcriptomics.Cell Syst. 2024 May 15;15(5):411-424.e9. doi: 10.1016/j.cels.2024.04.004. Cell Syst. 2024. PMID: 38754365
-
RNA velocity-current challenges and future perspectives.Mol Syst Biol. 2021 Aug;17(8):e10282. doi: 10.15252/msb.202110282. Mol Syst Biol. 2021. PMID: 34435732 Free PMC article. Review.
-
Interpretable generative deep learning: an illustration with single cell gene expression data.Hum Genet. 2022 Sep;141(9):1481-1498. doi: 10.1007/s00439-021-02417-6. Epub 2022 Jan 6. Hum Genet. 2022. PMID: 34988661 Free PMC article. Review.
Cited by
-
Applications of single-cell RNA sequencing in spermatogenesis and molecular evolution.Zool Res. 2024 May 18;45(3):575-585. doi: 10.24272/j.issn.2095-8137.2024.010. Zool Res. 2024. PMID: 38766742 Free PMC article. Review.
-
Biologically informed NeuralODEs for genome-wide regulatory dynamics.Genome Biol. 2024 May 21;25(1):127. doi: 10.1186/s13059-024-03264-0. Genome Biol. 2024. PMID: 38773638 Free PMC article.
-
Dissection and integration of bursty transcriptional dynamics for complex systems.Proc Natl Acad Sci U S A. 2024 Apr 30;121(18):e2306901121. doi: 10.1073/pnas.2306901121. Epub 2024 Apr 26. Proc Natl Acad Sci U S A. 2024. PMID: 38669186 Free PMC article.
-
TGF-β signaling redirects Sox11 gene regulatory activity to promote partial EMT and collective invasion of oncogenically transformed intestinal organoids.Oncogenesis. 2025 May 20;14(1):17. doi: 10.1038/s41389-025-00560-7. Oncogenesis. 2025. PMID: 40393982 Free PMC article.
-
RVINN: a flexible modeling for inferring dynamic transcriptional and post-transcriptional regulation using physics-informed neural networks.Bioinformatics. 2025 Jul 1;41(Supplement_1):i561-i570. doi: 10.1093/bioinformatics/btaf180. Bioinformatics. 2025. PMID: 40662812 Free PMC article.
