

Discrimination of cell-intrinsic and environment-dependent effects of natural genetic variation on Kupffer cell epigenomes and transcriptomes
- PMID: 37735593
- PMCID: PMC10602851
- DOI: 10.1038/s41590-023-01631-w
Discrimination of cell-intrinsic and environment-dependent effects of natural genetic variation on Kupffer cell epigenomes and transcriptomes
Abstract
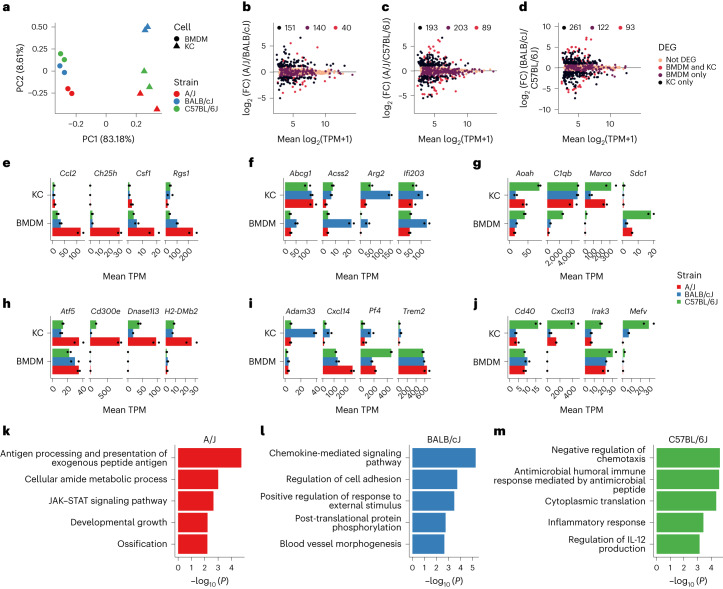
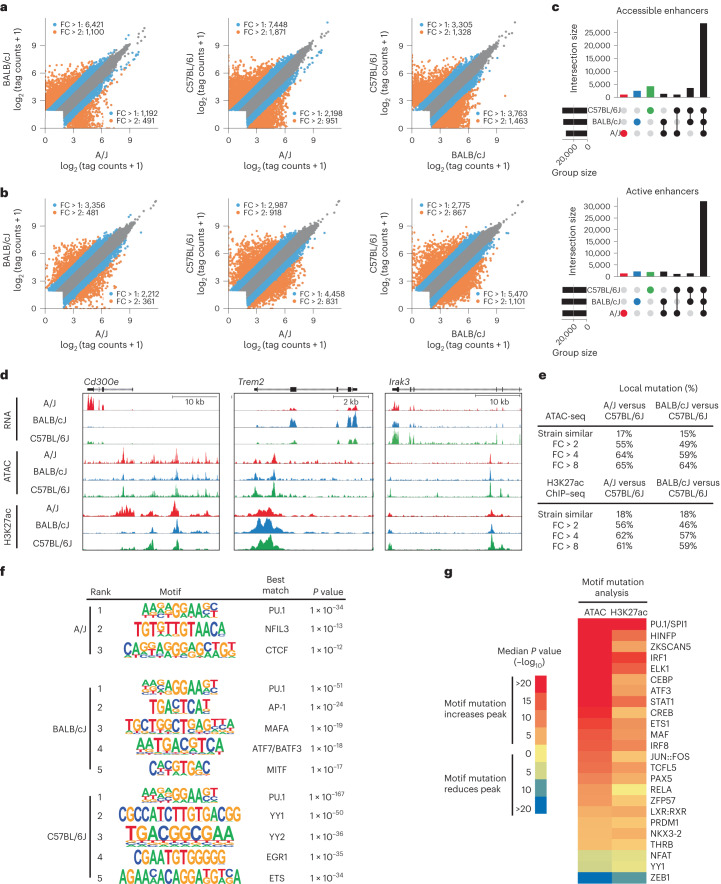
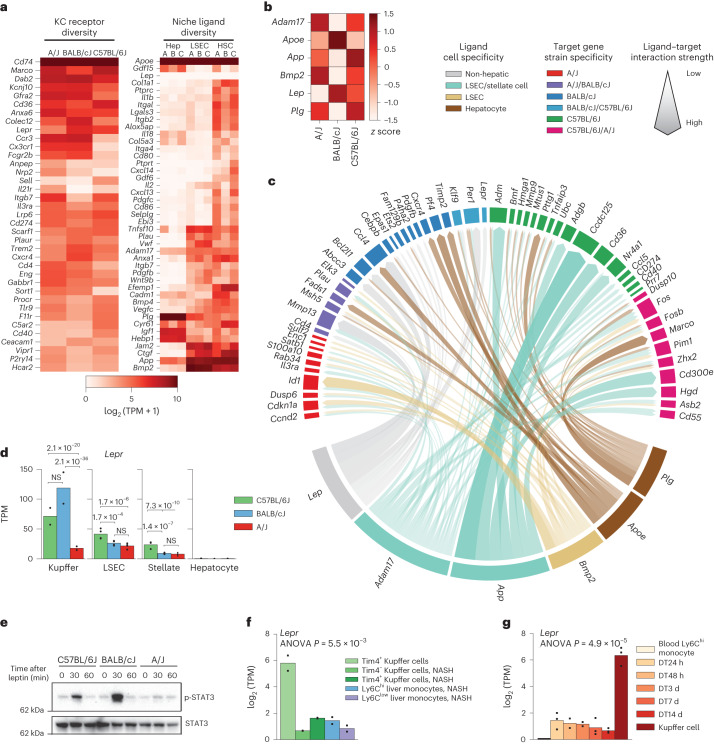
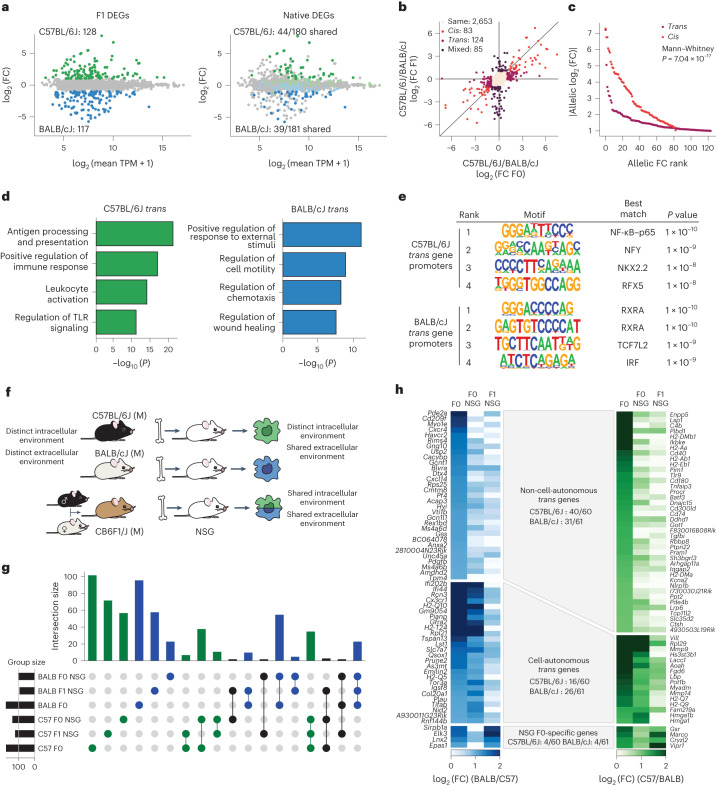
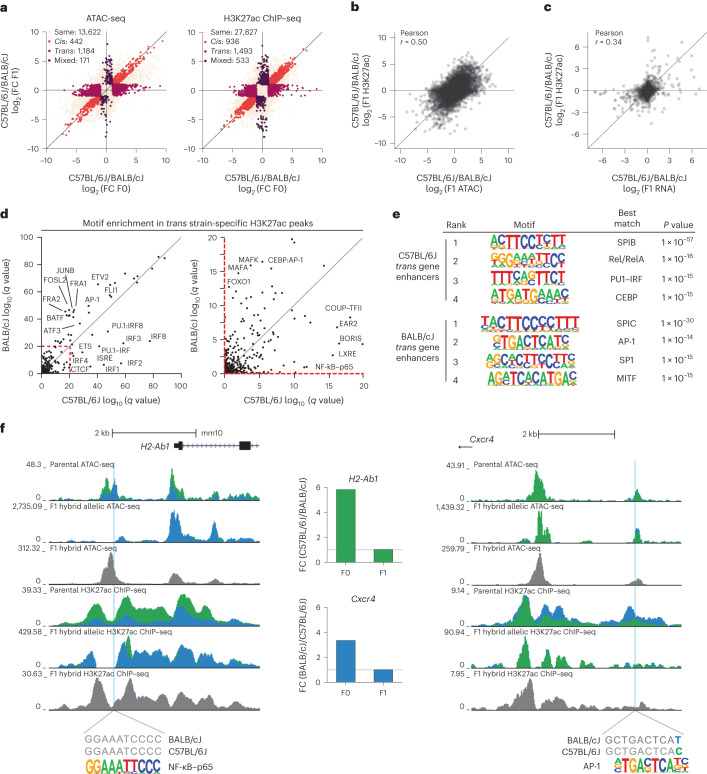
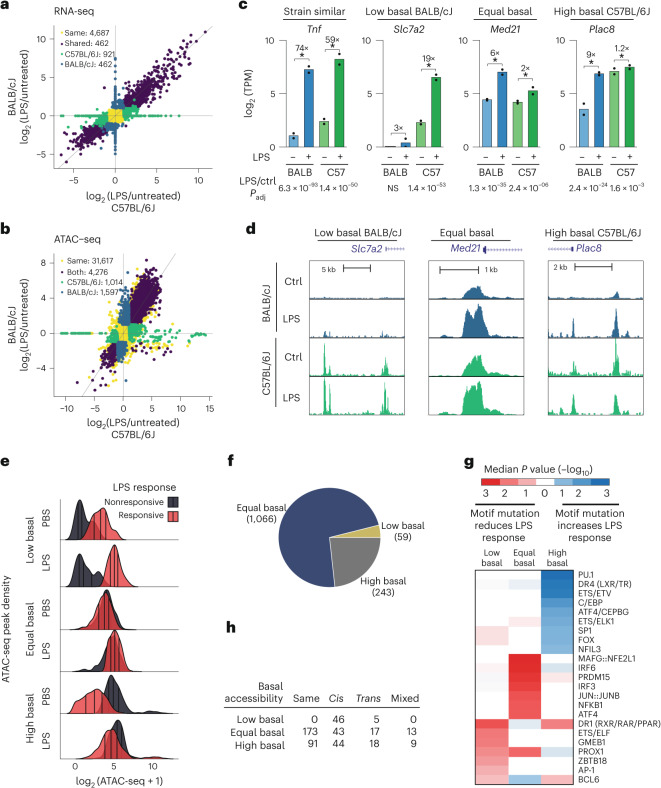
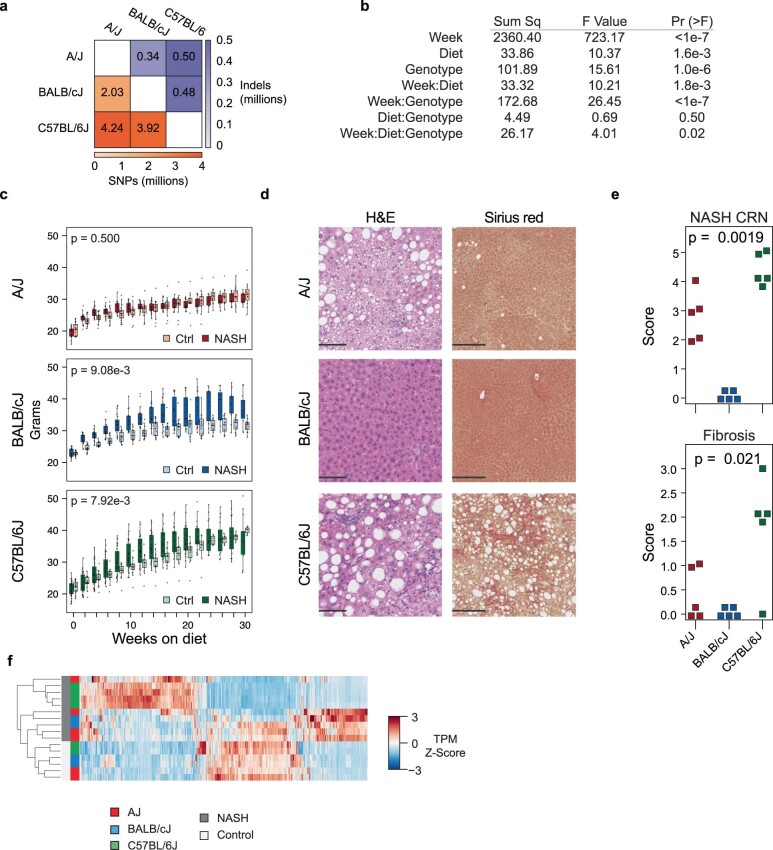
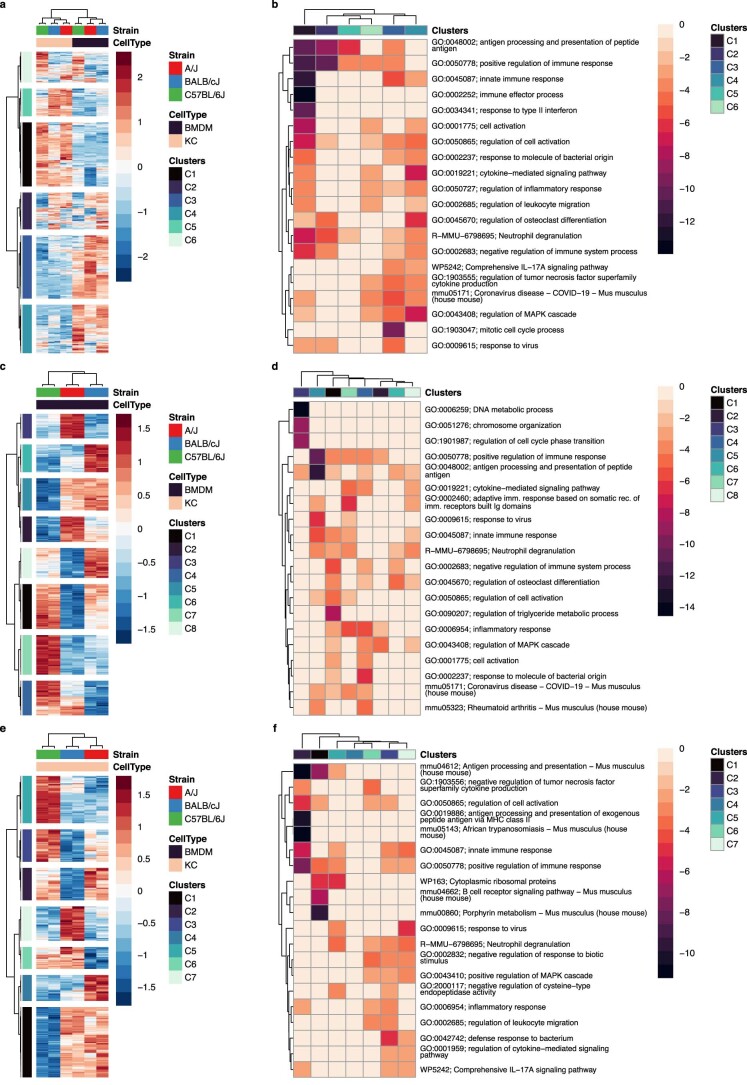
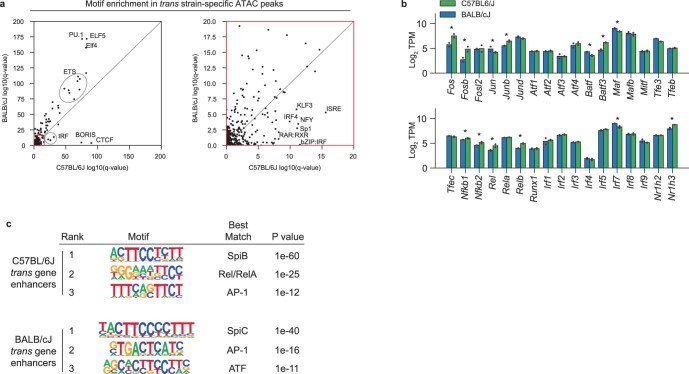
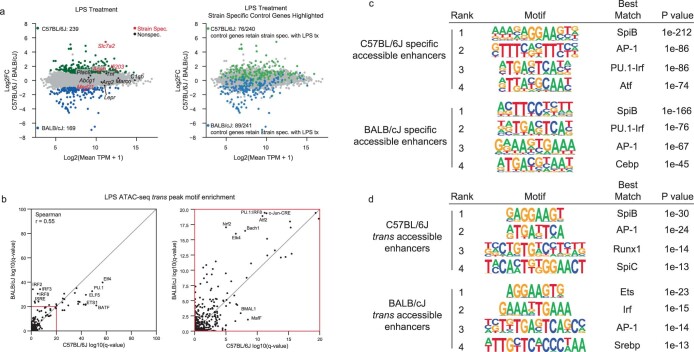
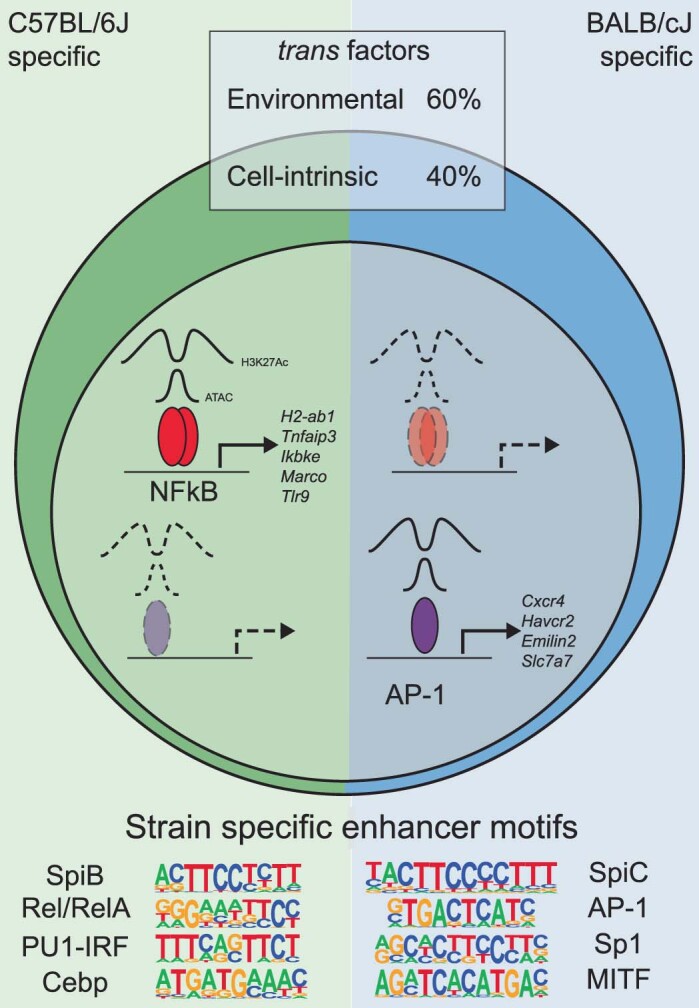
Noncoding genetic variation drives phenotypic diversity, but underlying mechanisms and affected cell types are incompletely understood. Here, investigation of effects of natural genetic variation on the epigenomes and transcriptomes of Kupffer cells derived from inbred mouse strains identified strain-specific environmental factors influencing Kupffer cell phenotypes, including leptin signaling in Kupffer cells from a steatohepatitis-resistant strain. Cell-autonomous and non-cell-autonomous effects of genetic variation were resolved by analysis of F1 hybrid mice and cells engrafted into an immunodeficient host. During homeostasis, non-cell-autonomous trans effects of genetic variation dominated control of Kupffer cells, while strain-specific responses to acute lipopolysaccharide injection were dominated by actions of cis-acting effects modifying response elements for lineage-determining and signal-dependent transcription factors. These findings demonstrate that epigenetic landscapes report on trans effects of genetic variation and serve as a resource for deeper analyses into genetic control of transcription in Kupffer cells and macrophages in vitro.
© 2023. The Author(s).
Conflict of interest statement
C.K.G. is a cofounder, equity holder and member of the Scientific Advisory Board of Asteroid Therapeutics. The other authors declare no competing interests.
Figures
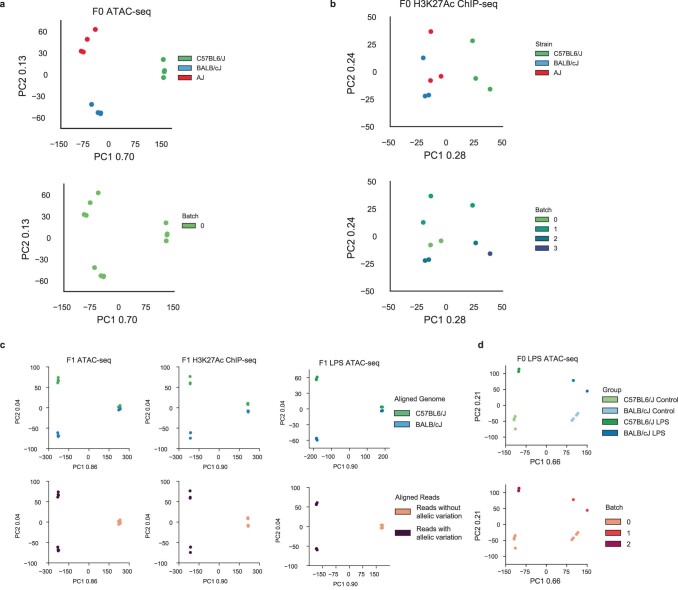
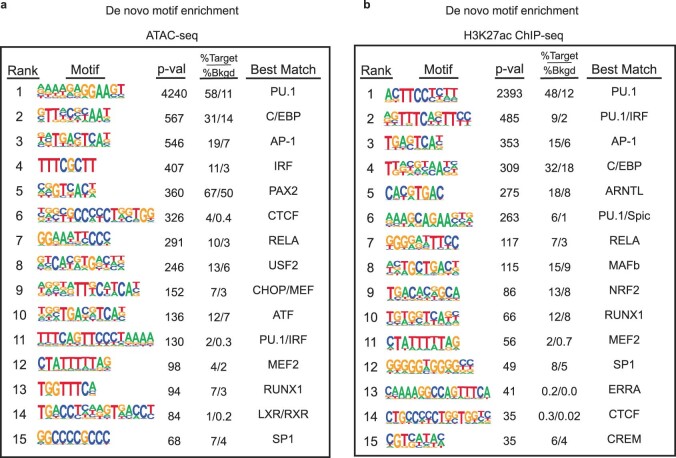
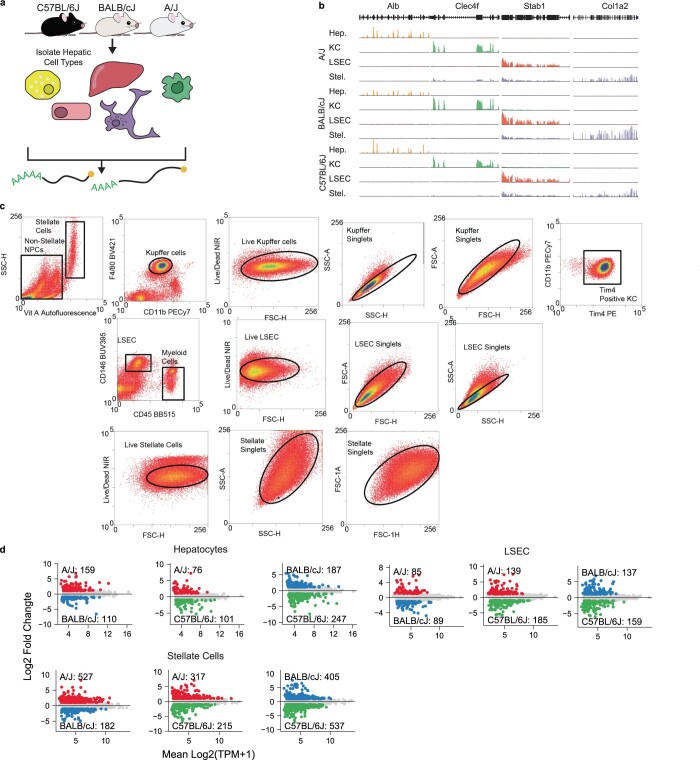
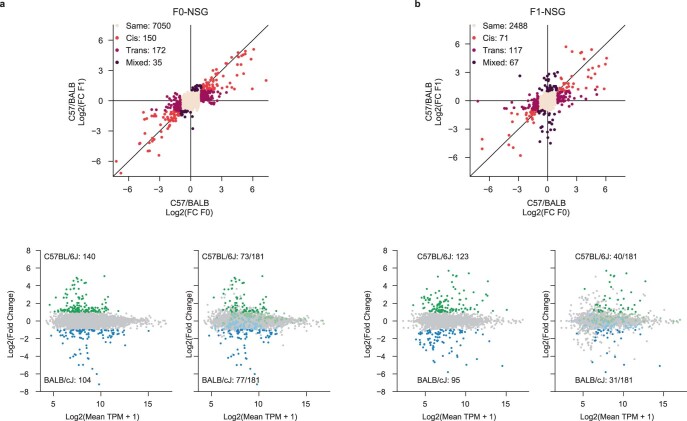
References
MeSH terms
Grants and funding
- T32 DK007044/DK/NIDDK NIH HHS/United States
- P30 DK078392/DK/NIDDK NIH HHS/United States
- P30 DK120515/DK/NIDDK NIH HHS/United States
- T32 DK007541/DK/NIDDK NIH HHS/United States
- T32 DK007202/DK/NIDDK NIH HHS/United States
- P01 HL147835/HL/NHLBI NIH HHS/United States
- T32 CA009523/CA/NCI NIH HHS/United States
- T32 GM007198/GM/NIGMS NIH HHS/United States
- R01 DK091183/DK/NIDDK NIH HHS/United States
- R37 AA020703/AA/NIAAA NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- F30 DK124980/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
