

PPP1R12C Promotes Atrial Hypocontractility in Atrial Fibrillation
- PMID: 37737016
- PMCID: PMC10616980
- DOI: 10.1161/CIRCRESAHA.123.322516
PPP1R12C Promotes Atrial Hypocontractility in Atrial Fibrillation
Abstract
Background: Atrial fibrillation (AF)-the most common sustained cardiac arrhythmia-increases thromboembolic stroke risk 5-fold. Although atrial hypocontractility contributes to stroke risk in AF, the molecular mechanisms reducing myofilament contractile function remain unknown. We tested the hypothesis that increased expression of PPP1R12C (protein phosphatase 1 regulatory subunit 12C)-the PP1 (protein phosphatase 1) regulatory subunit targeting MLC2a (atrial myosin light chain 2)-causes hypophosphorylation of MLC2a and results in atrial hypocontractility.
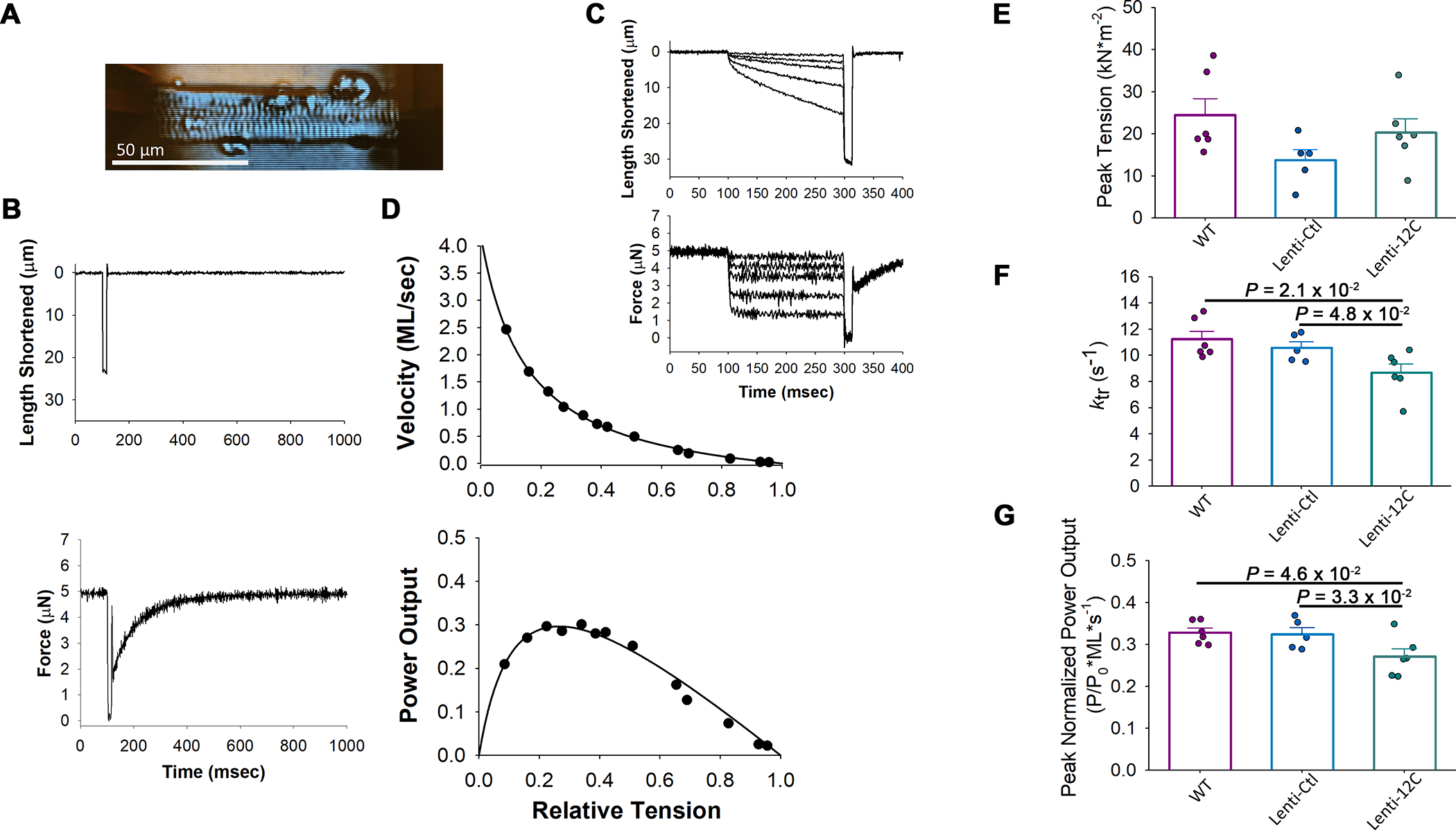
Methods: Right atrial appendage tissues were isolated from human patients with AF versus sinus rhythm controls. Western blots, coimmunoprecipitation, and phosphorylation studies were performed to examine how the PP1c (PP1 catalytic subunit)-PPP1R12C interaction causes MLC2a dephosphorylation. In vitro studies of pharmacological MRCK (myotonic dystrophy kinase-related Cdc42-binding kinase) inhibitor (BDP5290) in atrial HL-1 cells were performed to evaluate PP1 holoenzyme activity on MLC2a. Cardiac-specific lentiviral PPP1R12C overexpression was performed in mice to evaluate atrial remodeling with atrial cell shortening assays, echocardiography, and AF inducibility with electrophysiology studies.
Results: In human patients with AF, PPP1R12C expression was increased 2-fold versus sinus rhythm controls (P=2.0×10-2; n=12 and 12 in each group) with >40% reduction in MLC2a phosphorylation (P=1.4×10-6; n=12 and 12 in each group). PPP1R12C-PP1c binding and PPP1R12C-MLC2a binding were significantly increased in AF (P=2.9×10-2 and 6.7×10-3, respectively; n=8 and 8 in each group). In vitro studies utilizing drug BDP5290, which inhibits T560-PPP1R12C phosphorylation, demonstrated increased PPP1R12C binding with both PP1c and MLC2a and dephosphorylation of MLC2a. Mice treated with lentiviral PPP1R12C vector demonstrated a 150% increase in left atrial size versus controls (P=5.0×10-6; n=12, 8, and 12), with reduced atrial strain and atrial ejection fraction. Pacing-induced AF in mice treated with lentiviral PPP1R12C vector was significantly higher than in controls (P=1.8×10-2 and 4.1×10-2, respectively; n=6, 6, and 5).
Conclusions: Patients with AF exhibit increased levels of PPP1R12C protein compared with controls. PPP1R12C overexpression in mice increases PP1c targeting to MLC2a and causes MLC2a dephosphorylation, which reduces atrial contractility and increases AF inducibility. These findings suggest that PP1 regulation of sarcomere function at MLC2a is a key determinant of atrial contractility in AF.
Keywords: arrhythmias, cardiac; atrial fibrillation; myosin light chains; protein phosphatase 1; stroke volume.
Conflict of interest statement
Figures







Update of
-
Myosin Light Chain Dephosphorylation by PPP1R12C Promotes Atrial Hypocontractility in Atrial Fibrillation.bioRxiv [Preprint]. 2023 Apr 20:2023.04.19.537590. doi: 10.1101/2023.04.19.537590. bioRxiv. 2023. Update in: Circ Res. 2023 Oct 13;133(9):758-771. doi: 10.1161/CIRCRESAHA.123.322516. PMID: 37131731 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL062426/HL/NHLBI NIH HHS/United States
- R01 HL151508/HL/NHLBI NIH HHS/United States
- R38 HL155729/HL/NHLBI NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- R01 HL136389/HL/NHLBI NIH HHS/United States
- R01 HL091947/HL/NHLBI NIH HHS/United States
- F30 HL145980/HL/NHLBI NIH HHS/United States
- R01 HL131517/HL/NHLBI NIH HHS/United States
- R01 HL089598/HL/NHLBI NIH HHS/United States
- R01 HL128468/HL/NHLBI NIH HHS/United States
- R01 HL138737/HL/NHLBI NIH HHS/United States
- R01 HL158634/HL/NHLBI NIH HHS/United States
- I01 BX004268/BX/BLRD VA/United States
- R01 HL147108/HL/NHLBI NIH HHS/United States
- T32 DK101001/DK/NIDDK NIH HHS/United States
- I01 BX004918/BX/BLRD VA/United States
- R01 HL148785/HL/NHLBI NIH HHS/United States
- R01 HL117641/HL/NHLBI NIH HHS/United States
- R01 HL163277/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
