

Induced pluripotent stem cells: ex vivo models for human diseases due to mitochondrial DNA mutations
- PMID: 37737178
- PMCID: PMC10515435
- DOI: 10.1186/s12929-023-00967-7
Induced pluripotent stem cells: ex vivo models for human diseases due to mitochondrial DNA mutations
Abstract
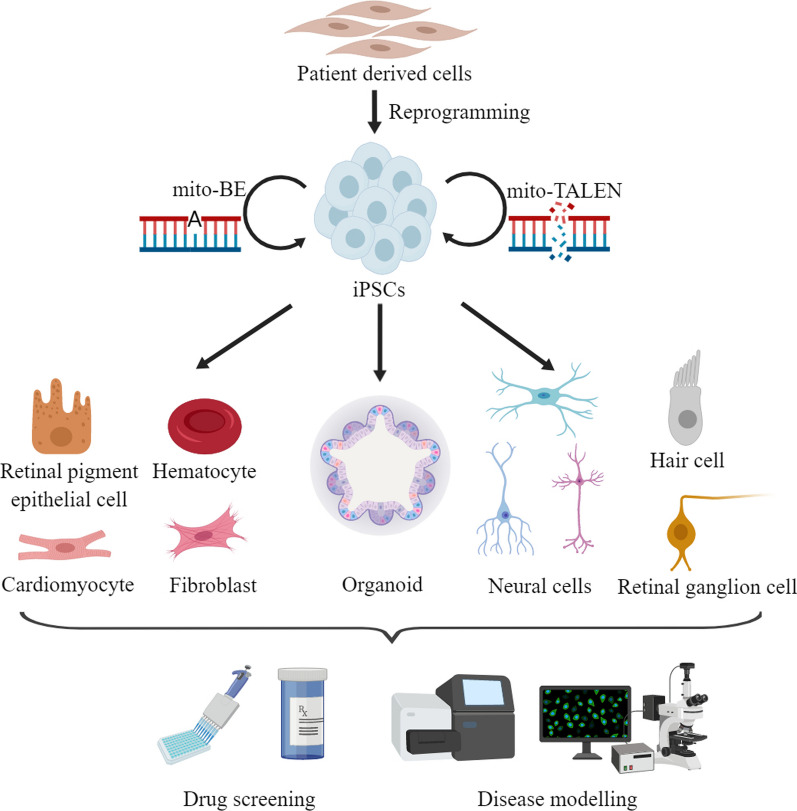
Mitochondria are essential organelles for cellular metabolism and physiology in eukaryotic cells. Human mitochondria have their own genome (mtDNA), which is maternally inherited with 37 genes, encoding 13 polypeptides for oxidative phosphorylation, and 22 tRNAs and 2 rRNAs for translation. mtDNA mutations are associated with a wide spectrum of degenerative and neuromuscular diseases. However, the pathophysiology of mitochondrial diseases, especially for threshold effect and tissue specificity, is not well understood and there is no effective treatment for these disorders. Especially, the lack of appropriate cell and animal disease models has been significant obstacles for deep elucidating the pathophysiology of maternally transmitted diseases and developing the effective therapy approach. The use of human induced pluripotent stem cells (iPSCs) derived from patients to obtain terminally differentiated specific lineages such as inner ear hair cells is a revolutionary approach to deeply understand pathogenic mechanisms and develop the therapeutic interventions of mitochondrial disorders. Here, we review the recent advances in patients-derived iPSCs as ex vivo models for mitochondrial diseases. Those patients-derived iPSCs have been differentiated into specific targeting cells such as retinal ganglion cells and eventually organoid for the disease modeling. These disease models have advanced our understanding of the pathophysiology of maternally inherited diseases and stepped toward therapeutic interventions for these diseases.
Keywords: Maternally inherited diseases; Mitochondria; iPSCs; mtDNA mutations.
© 2023. National Science Council of the Republic of China (Taiwan).
Conflict of interest statement
All authors have no proprietary or commercial interest in any of materials discussed in this article.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
